IVD-Charter
In-vitro diagnostica (IVD’s) zijn medische hulpmiddelen voor het in-vitro onderzoek van specimens die afkomstig zijn van het menselijk lichaam, met het doel informatie te verschaffen over een of meer van de volgende elementen:


Gerelateerde artikels

Jeroen Poels, FAGG: 'IVDR moet positief verhaal worden voor iedereen' (deel 2/2)
Vorige week spraken we met Jeroen Poels, expert in-vitro diagnostica bij het FAGG, over de strengere vereisten onder de IVDR en de administratieve impact daarvan op bedrijven. Vandaag zoomen we uit en kijken we naar de betekenis van de IVDR voor de verschillende stakeholders.
Mijnheer Poels, vorige week sloten we af met de administratieve druk die de IVDR met zich meebrengt voor bedrijven. Meer administratie betekent ook hogere kosten voor die bedrijven. Merken jullie dat met het FAGG?
De overgang naar de nieuwe wetgeving is inderdaad niet te onderschatten voor IVD-bedrijven. Voor "medical device"- of MD-bedrijven is de regelgeving ook veranderd met de komst van de Medical Device Regulation (MDR), maar de wijzigingen zijn minder ingrijpend dan die onder de IVDR.
'IVD-bedrijven zijn vaak kmo's en beschikken niet over enorme middelen.'
Bovendien zijn heel wat IVD-bedrijven kmo's die niet over dezelfde middelen beschikken als de vaak grote MD-fabrikanten. Europa probeert daar rekening mee te houden, bijvoorbeeld met de SME Relief Package (2023) en met een EU-strategie specifiek voor start-ups en scale-ups (2025).
Door de IVDR later in werking te laten treden en door in extra overgangsperiodes te voorzien, wil Europa alle betrokken stakeholders voldoende tijd geven om zich in orde te stellen.
De druk op IVD-bedrijven blijft evenwel een feit. De scherpere eisen rond klinisch bewijs en de controles door notified bodies en eventueel ook Europese referentielaboratoria verhogen de kosten voor bedrijven onvermijdelijk. We horen dan ook dat IVD-fabrikanten hun productportfolio kritisch herevalueren - welke producten willen ze "meenemen" naar het IVDR-kader en welke zetten ze stop?
De zwaardere IVDR maakt innoveren onvermijdelijk nog duurder dan het al was. Vrezen jullie dat de IVDR een rem kan zetten op vernieuwing?
Dat valt vandaag moeilijk te voorspellen, maar ik hoop alvast van niet. Europa wil het in elk geval niet zover laten komen en heeft een werkgroep opgericht om de procedures voor "breakthrough innovation" te versnellen.
Het FAGG trekt, als sterk gewaardeerde autoriteit binnen de EU, volop mee aan die kar.
Het FAGG is inderdaad een sterk internationaal merk in Europa. Dat blijkt ook duidelijk uit de zeer actieve rol die jullie spelen in onder andere de Medical Device Coordination Group (MDCG).
Onze administratie is inderdaad zeer actief, en dat in alle 13 werkgroepen van de MDCG. Die werkgroepen behandelen alle aspecten die relevant zijn voor de implementatie van de IVDR en de MDR, zoals aanmelding en controle van aangemelde instanties, vigilantie, marktcontrole, klinische en performantiestudies, etc.
'Bevoegde instanties van andere lidstaten doen geregeld een beroep op de Belgische expertise rond IVD's.'
Eén werkgroep focust specifiek op de homogene interpretatie en implementatie met betrekking tot IVD's en verleent de andere 12 groepen advies over IVD-specifieke topics. Sinds 2022 heeft de IVD-werkgroep liefst 16 guidance documenten gepubliceerd. Met het FAGG hebben we aan elk van die 16 documenten meegewerkt, vaak in een leidende rol. De bevoegde instanties van andere EU-lidstaten erkennen die expertise en vragen geregeld onze opinie over IVD-topics.
Zelf ben ik trouwens recent covoorzitter geworden van de taskforce voor orphan IVD's - een orphan IVD is een test voor een zeer kleine groep patiënten. Met die taskforce onderzoeken we hoe we wees-IVD's makkelijker en sneller beschikbaar kunnen maken in Europa.
We hebben het al uitgebreid gehad over de extra druk die de IVDR met zich meebrengt voor bedrijven. Hoe zit het met de druk op jullie administratie?
De EUDAMED-databank moet op termijn een deel van de administratieve last voor ons helpen opvangen, maar dat is vandaag nog niet het geval. Intussen zijn er heel wat taken uitgebreid en hebben we nieuwe taken gekregen. Dus ja, de IVDR brengt dus ook voor ons extra druk met zich mee, zeker op korte termijn.
Tezelfdertijd zijn er belangrijke positieve signalen. Lang was er de terechte vrees dat de capaciteit aan aangemelde instanties niet zou volstaan voor de IVDR - het aantal IVD's dat een conformiteitsbeoordeling moet ondergaan, is toegenomen van 20 naar 80% (zie ook deel 1 van het interview). Maar dat probleem lijkt zich op te lossen.
Van de 17 notified bodies voor de IVDR zijn er momenteel slechts 2 die geen nieuwe klanten meer aannemen, zo blijkt uit een recente enquête. Eén van de nieuwste NB's op de lijst is trouwens SGS Belgium; daarmee heeft ook ons land nu een aangemelde instantie voor de IVDR, wat uiteraard goed nieuws is voor fabrikanten in België!

Verder zien we het aantal aanvragen voor prestatiestudies toenemen. Vaak gaat het om combinatiestudies met een geneesmiddel. De IVD-test dient dan om patiënten voor de trial te selecteren en wordt later eventueel als companion diagnostic gecommercialiseerd. Er loopt momenteel een groot Europees project, COMBINE, om de aanvragen van combinatiestudies te vereenvoudigen en harmoniseren.
Die signalen zijn erg belangrijk. De IVDR moet tenslotte een positief verhaal worden.
Absoluut, in de eerste plaats voor individuele patiënten en de volksgezondheid.
De potentiële voordelen van de IVDR voor patiënten zijn overduidelijk: hoe performanter en veiliger medische hulpmiddelen voor in-vitro diagnostiek, hoe beter voor de patiënt. En dankzij de betere traceerbaarheid kunnen hulpmiddelen zeer gericht opgespoord worden als er toch iets fout mee zou lopen.
'Hoe performanter en veiliger IVD's zijn, hoe beter voor de patiënt.'
Alle maatregelen binnen de IVDR dragen daar in meer of mindere mate aan bij. Het is onvermijdelijk dat bij een dergelijke grote wijziging aan de regelgeving er ook heel wat aandachtspunten zijn. Maar die mogen op zich geen reden vormen om de hele hervorming in vraag te stellen.
Het is zaak om de IVDR continu en nauwgezet te evalueren en te durven bijsturen waar nodig: wat kan eenvoudiger, welke vereisten bieden te weinig meerwaarde en kunnen we dus bijstellen of afschaffen, etc. Europa gaat actief op zoek naar die feedback - zo is eind maart een publieke consultatie bij stakeholders afgerond over de IVDR en de MDR, waarvan de resultaten momenteel worden verwerkt. En ook met het FAGG blijven we feedback verzamelen bij de betrokken stakeholders in België.

Jeroen Poels, FAGG: 'Bedrijven kunnen onze voelsprieten zijn' (deel 1/2)
De IVDR is sinds eind vorige maand 3 jaar van kracht. Hoe ervoer het FAGG de nieuwe regelgeving tot nu toe? Wat loopt goed en wat kan beter? We praatten met Jeroen Poels, expert voor IVD's binnen het FAGG. "Hoe vroeger we mogelijke problemen met de IVDR opmerken, hoe meer we eraan kunnen doen. Maar daarvoor hebben we de input van bedrijven nodig."
De Europese In Vitro Diagnostics Regulation, kortweg IVDR, is van toepassing sinds 26 mei 2022. De verordening verving de In Vitro Diagnostics Directive (IVDD). Doel? De veiligheid en kwaliteit van in-vitro diagnostica verbeteren, de transparantie over en traceerbaarheid van IVD's verhogen en de implementatie van de wetgeving binnen de EU harmoniseren.
In een tweedelig interview fileert Jeroen Poels de IVDR. Hij is expert medische hulpmiddelen voor in-vitrodiagnostiek bij het Federaal Agentschap voor Geneesmiddelen en Gezondheidsproducten (FAGG), dat toeziet op de veiligheid, kwaliteit en doeltreffendheid van medische hulpmiddelen op de Belgische markt.
Mijnheer Poels, laat ons beginnen bij het begin. Waarom was de IVDD aan vervanging toe?
De IVDD dateerde van 1998, een tijd waarin de diagnostische mogelijkheden veel beperkter waren dan nu. Er was toen bijvoorbeeld nog bijna geen sprake van genetische tests, point-of-care tests, IVD-software, noem maar op. Om al die nieuwe, vaak complexe mogelijkheden juist te omkaderen en te anticiperen op nieuwe mogelijkheden, moest de regelgeving volledig herdacht worden.
Bij het uittekenen van een nieuw kader, koos Europa zeer bewust voor een verordening in plaats van een richtlijn. Een richtlijn kunnen lidstaten elk op hun manier omzetten in nationale wetgeving. Dat biedt flexibiliteit, maar de uitdagingen rond veilige, kwalitatieve diagnostische hulpmiddelen overstijgen de landsgrenzen. Vandaar de keuze voor een verordening, die elke lidstaat quasi op dezelfde manier moet toepassen.
Wat zijn de belangrijkste nieuwigheden ten opzichte van de IVDD?
De IVDR telt 160 pagina's, de IVDD telde er 37. Verschillen zijn er dus genoeg. De belangrijkste hebben te maken met de vereisten waaraan IVD's moeten voldoen en de controle daarop.
Vroeger, onder de IVDD, werd een beperkt aantal IVD's als "hoog risico" gecatalogeerd. Denk aan hiv- of hepatitistests. Die IVD's moesten een conformiteitsbeoordeling ondergaan door een notified body (NB) of aangemelde instantie - dat ging om ongeveer 20% van alle IVD's - vooraleer ze op de markt konden komen. Voor de andere 80% was zelfcertificering door de fabrikant voldoende.
'Vroeger moest 20% van alle IVD's gecontroleerd worden door een notified body, vandaag gaat het om 80%.'
Onder de IVDR hebben alle IVD's een risicoklasse gekregen (van laag naar hoog, A tot en met D) en is de verhouding omgekeerd: circa 80% van alle IVD's moet nu gecontroleerd worden door een notified body, tegenover 20% vroeger. Enkel voor niet-steriele IVD's van klasse A is zelfcertificering nog toegelaten, bijvoorbeeld kleurreagentia.
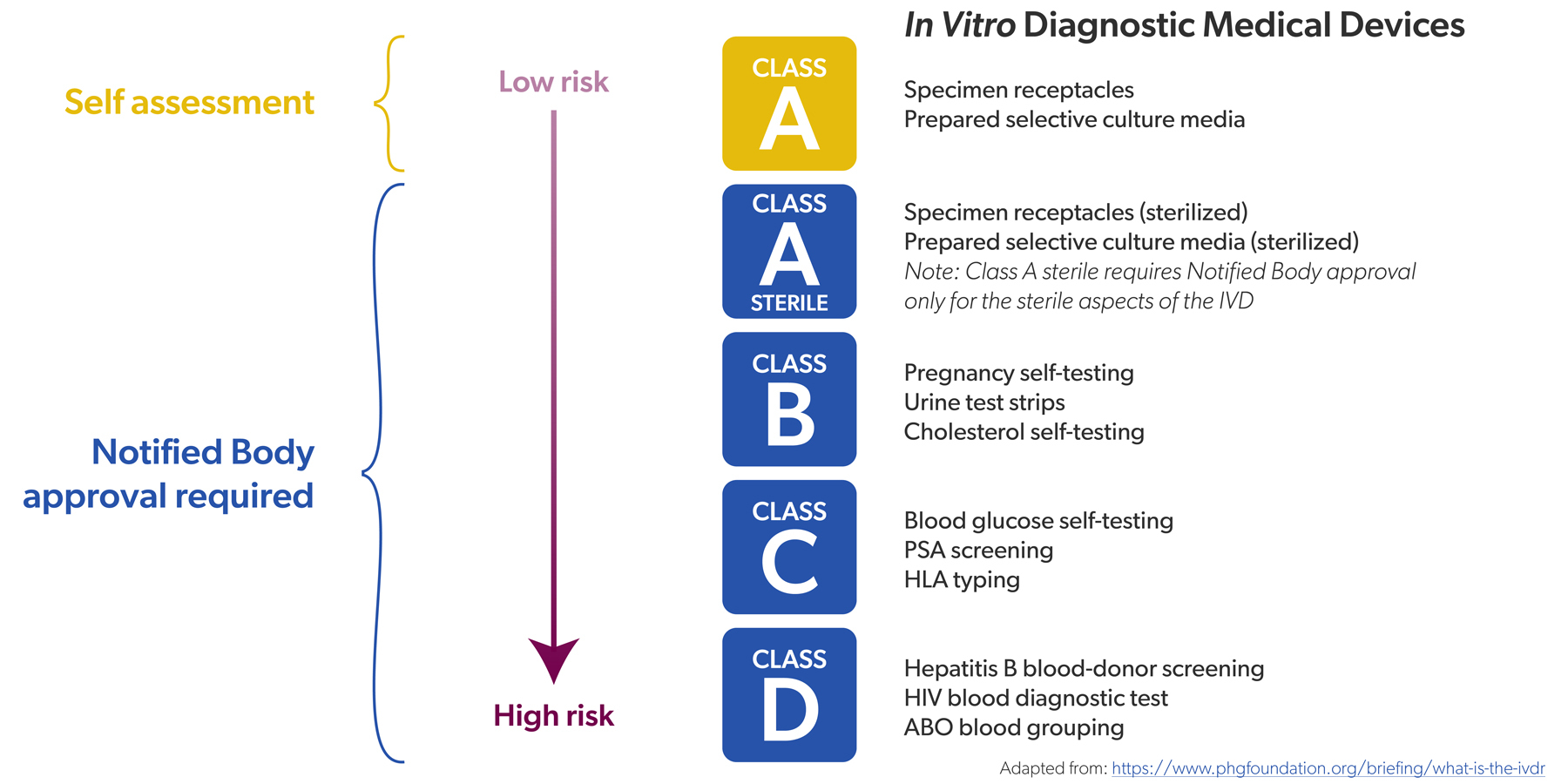
De conformiteitsbeoordelingen zijn ook strenger, net als het toezicht op de NB's die ze uitvoeren. Voor hoogrisico-IVD's zijn er nu trouwens extra actoren die de conformiteit mee beoordelen, zoals het IVD Expert Panel en de Europese referentielaboratoria. Hun expertise is een belangrijke troef.
Wat houdt zo'n strengere conformiteitsbeoordeling concreet in?
De vereisten op het vlak van productveiligheid, identificatie en traceerbaarheid zijn serieus aangescherpt. Ook op het vlak van prestaties ligt de lat veel hoger. Vooraleer een IVD in de handel gebracht kan worden, wordt nu veel meer klinisch bewijs gevraagd van de werking van het hulpmiddel.
Onder de IVDD volstond een verklaring van de fabrikant dat er een prestatiestudie zou worden uitgevoerd. De IVDR legt de bewijslast veel hoger, zeker voor studies met een hoger risico voor deelnemende patiënten. Wie zo'n studie wil uitvoeren, moet bovendien op voorhand goedkeuring krijgen van zowel een ethisch comité als de bevoegde autoriteit.
'Recent is een Europees pilootproject gestart om de aanvraagprocedure voor multi-country studies te vereenvoudigen.'
Tezelfdertijd zoekt Europa manieren om de administratieve overlast te beperken. In dat kader is recent een pilootproject gestart waarbij sponsors die een studie in meerdere lidstaten willen uitvoeren, een enkelvoudige aanvraag kunnen indienen. De beoordeling van zo'n aanvraag zal gecoördineerd verlopen onder leiding van één bevoegde autoriteit, in plaats van door elke lidstaat afzonderlijk.
Over administratie (over)last gesproken: MedTech Europe waarschuwt dat de extra vereisten bedrijven voor moeilijkheden plaatsen, waardoor bepaalde IVD's onbeschikbaar dreigen te worden in de EU. In België zien we momenteel nog geen problemen, maar we volgen dit wel op. Hoe kijkt het FAGG hiernaar?
Als overheid zijn we zeker niet blind voor die problematiek. Integendeel, we zijn ons goed bewust van de mogelijke risico's en houden de situatie nauwgezet in de gaten. Ook Europa is zich daar bewust van. Daarom verlengde het bijvoorbeeld de overgangsperiodes voor IVD's die al onder de IVDD op de markt werden gebracht.
Om een beter zicht te krijgen op mogelijke tekorten, voerde het recent ook de verplichting in voor fabrikanten om bepaalde onderbrekingen of stopzettingen op voorhand te melden aan autoriteiten en klanten.
In België zien we inderdaad geen acute problemen. We hebben momenteel enkel weet van een belangrijke syfilistest van een Aziatisch bedrijf die niet meer op de markt zal komen in Europa. Maar België is in de eerste plaats een distributieland - fabrikanten van IVD's zijn vooral in andere landen actief. Dat maakt dat we meestal niet als eersten op de hoogte zijn als een fabrikant beslist om de productie van een test stop te zetten.
Onze oproep aan beMedTech en aan jullie leden is daarom duidelijk: als jullie iets voelen borrelen, nog voor het een echt probleem wordt, reik daar dan alsjeblieft over uit naar ons. Dan zullen we er alles aan doen om samen oplossingen te zoeken.
Het is zoals met gezondheidsproblemen: hoe vroeger je erbij bent, hoe meer kans je hebt om escalatie te vermijden. Jullie leden zijn de voelsprieten die we nodig hebben om potentiële problemen in een zo vroeg mogelijk stadium te detecteren.

Prof. Cavalier: ‘Investeren in laboratoriumgeneeskunde is essentieel voor kwaliteitsvolle zorg'
Tijdens EuroMedLab 2025 spraken we met prof. dr. Etienne Cavalier, professor Klinische Chemie aan ULiège, diensthoofd Klinische Chemie aan CHU de Liège en voorzitter van de Royal Belgian Society of Laboratory Medicine. Welke maatregelen rond laboratoriumgeneeskunde zou hij nemen als hij federaal minister van Volksgezondheid was?
“Mijn absolute prioriteit zou zijn om sneller toegang te verlenen tot nieuwe tests met een hoge toegevoegde waarde. Nu duurt het vaak jaren vooraleer er terugbetaling is, als die er al komt. Zo worden de NT-proBNP- en BNP-tests nog steeds niet terugbetaald in België, terwijl ze in alle internationale richtlijnen staan. Die lange wachttijd is nefast voor de Belgische patiënt.”
'Terugverdieneffecten'
Net als prof. Mario Plebani (*) gelooft prof. Cavalier dat het geen kwestie van middelen is.
(*) Naar aanleiding van EuroMedLab 2025 spraken we ook met professor Mario Plebani, voorzitter van de European Federation of Clinical Chemistry and Laboratory Medicine. Dat interview lees je HIER.
“Eerst en vooral leveren veel nieuwe tests terugverdieneffecten op. Op korte termijn, doordat ze duurdere, vaak invasieve procedures vervangen. En op langere termijn, doordat er minder zorg nodig is dankzij de betere detectie. Helaas worden die effecten niet meegenomen bij de evaluatie om nieuwe tests wel of niet terug te betalen.”
“Daarnaast worden er in België nog steeds tests terugbetaald waarvan de klinische relevantie beperkt is. En bij sommige tests zouden we de voorgeschreven frequentie kunnen verminderen. Door voor dergelijke tests in beperkingen te voorzien, komt er eveneens budgettaire ruimte vrij”, aldus prof. Cavalier.
Expertise van laboratoriumprofessionals
Maar laboratoriumgeneeskunde draait natuurlijk niet om tests alleen. De expertise van laboratoriumprofessionas is minstens zo belangrijk. Die expertise beter benutten, zeker in klinische zorgpaden voor chronische aandoeningen zou een tweede prioriteit zijn van prof. Cavalier als hij minister van Volksgezondheid was.
“Labo’s hebben een cruciale rol te spelen bij zaken als vroegdetectie en risicostratificatie. Maar we nemen onze rol als specialist te weinig op – of krijgen daar te weinig ruimte voor.”
'Laboratoriumprofessionals zouden eigenlijk mee rond de tafel moeten zitten met de andere klinische specialisten betrokken in het interdisciplinaire zorgpad.'
“We leveren soms testresultaten af waar vervolgens te weinig mee gebeurt. En dat zijn gemiste kansen om de zorg voor de patiënt te verbeteren. We zouden als labo-expert eigenlijk mee rond de tafel moeten zitten met de andere klinische specialisten betrokken in het interdisciplinaire zorgpad.”
“Dat vraagt een andere kijk op laboratoriumgeneeskunde. Te veel stakeholders zien onze discipline nog vooral als een kost, terwijl goede diagnostiek net een investering is in kwalitatieve value-based zorg.”