De Europese In Vitro Diagnostics Regulation, kortweg IVDR, is van toepassing sinds 26 mei 2022. De verordening verving de In Vitro Diagnostics Directive (IVDD). Doel? De veiligheid en kwaliteit van in-vitro diagnostica verbeteren, de transparantie over en traceerbaarheid van IVD's verhogen en de implementatie van de wetgeving binnen de EU harmoniseren.
In een tweedelig interview fileert Jeroen Poels de IVDR. Hij is expert medische hulpmiddelen voor in-vitrodiagnostiek bij het Federaal Agentschap voor Geneesmiddelen en Gezondheidsproducten (FAGG), dat toeziet op de veiligheid, kwaliteit en doeltreffendheid van medische hulpmiddelen op de Belgische markt.
Mijnheer Poels, laat ons beginnen bij het begin. Waarom was de IVDD aan vervanging toe?
De IVDD dateerde van 1998, een tijd waarin de diagnostische mogelijkheden veel beperkter waren dan nu. Er was toen bijvoorbeeld nog bijna geen sprake van genetische tests, point-of-care tests, IVD-software, noem maar op. Om al die nieuwe, vaak complexe mogelijkheden juist te omkaderen en te anticiperen op nieuwe mogelijkheden, moest de regelgeving volledig herdacht worden.
Bij het uittekenen van een nieuw kader, koos Europa zeer bewust voor een verordening in plaats van een richtlijn. Een richtlijn kunnen lidstaten elk op hun manier omzetten in nationale wetgeving. Dat biedt flexibiliteit, maar de uitdagingen rond veilige, kwalitatieve diagnostische hulpmiddelen overstijgen de landsgrenzen. Vandaar de keuze voor een verordening, die elke lidstaat quasi op dezelfde manier moet toepassen.
Wat zijn de belangrijkste nieuwigheden ten opzichte van de IVDD?
De IVDR telt 160 pagina's, de IVDD telde er 37. Verschillen zijn er dus genoeg. De belangrijkste hebben te maken met de vereisten waaraan IVD's moeten voldoen en de controle daarop.
Vroeger, onder de IVDD, werd een beperkt aantal IVD's als "hoog risico" gecatalogeerd. Denk aan hiv- of hepatitistests. Die IVD's moesten een conformiteitsbeoordeling ondergaan door een notified body (NB) of aangemelde instantie - dat ging om ongeveer 20% van alle IVD's - vooraleer ze op de markt konden komen. Voor de andere 80% was zelfcertificering door de fabrikant voldoende.
'Vroeger moest 20% van alle IVD's gecontroleerd worden door een notified body, vandaag gaat het om 80%.'
Onder de IVDR hebben alle IVD's een risicoklasse gekregen (van laag naar hoog, A tot en met D) en is de verhouding omgekeerd: circa 80% van alle IVD's moet nu gecontroleerd worden door een notified body, tegenover 20% vroeger. Enkel voor niet-steriele IVD's van klasse A is zelfcertificering nog toegelaten, bijvoorbeeld kleurreagentia.
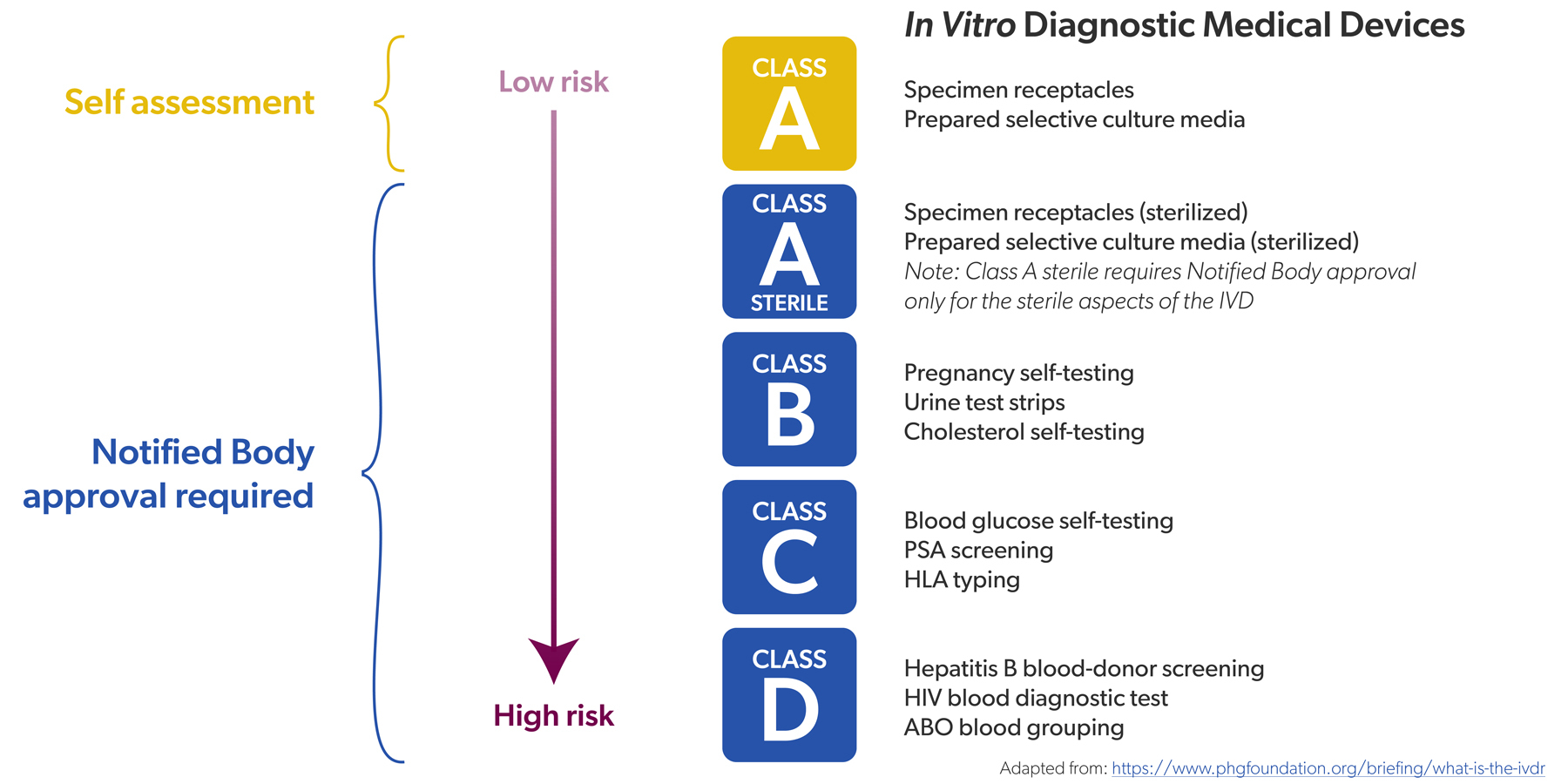
De conformiteitsbeoordelingen zijn ook strenger, net als het toezicht op de NB's die ze uitvoeren. Voor hoogrisico-IVD's zijn er nu trouwens extra actoren die de conformiteit mee beoordelen, zoals het IVD Expert Panel en de Europese referentielaboratoria. Hun expertise is een belangrijke troef.
Wat houdt zo'n strengere conformiteitsbeoordeling concreet in?
De vereisten op het vlak van productveiligheid, identificatie en traceerbaarheid zijn serieus aangescherpt. Ook op het vlak van prestaties ligt de lat veel hoger. Vooraleer een IVD in de handel gebracht kan worden, wordt nu veel meer klinisch bewijs gevraagd van de werking van het hulpmiddel.
Onder de IVDD volstond een verklaring van de fabrikant dat er een prestatiestudie zou worden uitgevoerd. De IVDR legt de bewijslast veel hoger, zeker voor studies met een hoger risico voor deelnemende patiënten. Wie zo'n studie wil uitvoeren, moet bovendien op voorhand goedkeuring krijgen van zowel een ethisch comité als de bevoegde autoriteit.
'Recent is een Europees pilootproject gestart om de aanvraagprocedure voor multi-country studies te vereenvoudigen.'
Tezelfdertijd zoekt Europa manieren om de administratieve overlast te beperken. In dat kader is recent een pilootproject gestart waarbij sponsors die een studie in meerdere lidstaten willen uitvoeren, een enkelvoudige aanvraag kunnen indienen. De beoordeling van zo'n aanvraag zal gecoördineerd verlopen onder leiding van één bevoegde autoriteit, in plaats van door elke lidstaat afzonderlijk.
Over administratie (over)last gesproken: MedTech Europe waarschuwt dat de extra vereisten bedrijven voor moeilijkheden plaatsen, waardoor bepaalde IVD's onbeschikbaar dreigen te worden in de EU. In België zien we momenteel nog geen problemen, maar we volgen dit wel op. Hoe kijkt het FAGG hiernaar?
Als overheid zijn we zeker niet blind voor die problematiek. Integendeel, we zijn ons goed bewust van de mogelijke risico's en houden de situatie nauwgezet in de gaten. Ook Europa is zich daar bewust van. Daarom verlengde het bijvoorbeeld de overgangsperiodes voor IVD's die al onder de IVDD op de markt werden gebracht.
Om een beter zicht te krijgen op mogelijke tekorten, voerde het recent ook de verplichting in voor fabrikanten om bepaalde onderbrekingen of stopzettingen op voorhand te melden aan autoriteiten en klanten.
In België zien we inderdaad geen acute problemen. We hebben momenteel enkel weet van een belangrijke syfilistest van een Aziatisch bedrijf die niet meer op de markt zal komen in Europa. Maar België is in de eerste plaats een distributieland - fabrikanten van IVD's zijn vooral in andere landen actief. Dat maakt dat we meestal niet als eersten op de hoogte zijn als een fabrikant beslist om de productie van een test stop te zetten.
Onze oproep aan beMedTech en aan jullie leden is daarom duidelijk: als jullie iets voelen borrelen, nog voor het een echt probleem wordt, reik daar dan alsjeblieft over uit naar ons. Dan zullen we er alles aan doen om samen oplossingen te zoeken.
Het is zoals met gezondheidsproblemen: hoe vroeger je erbij bent, hoe meer kans je hebt om escalatie te vermijden. Jullie leden zijn de voelsprieten die we nodig hebben om potentiële problemen in een zo vroeg mogelijk stadium te detecteren.