Le règlement européen sur les dispositifs médicaux de diagnostic in vitro, ou IVDR, est applicable depuis le 26 mai 2022. Il remplace la directive sur les dispositifs médicaux de diagnostic in vitro (IVDD). Son objectif ? Améliorer la sécurité et la qualité des dispositifs médicaux de diagnostic in vitro (DIV), renforcer la transparence et la traçabilité des DIV et harmoniser la mise en œuvre de la législation au sein de l'Union Européenne (UE)
Dans une interview en deux parties, Jeroen Poels décortique l'IVDR. Il est expert en dispositifs médicaux pour le diagnostic in vitro à l'Agence fédérale des médicaments et des produits de santé (AFMPS), qui veille à la sécurité, à la qualité et à l'efficacité des dispositifs médicaux sur le marché belge.
Monsieur Poels, commençons par le commencement. Pourquoi fallait-il remplacer la directive IVDD ?
L'IVDD datait de 1998, à une époque où les possibilités diagnostiques étaient beaucoup plus limitées qu'aujourd'hui. À l'époque, par exemple, les tests génétiques, les tests au chevet du patient (point of care tests ou POCT), les logiciels DIV, etc. n'existaient pratiquement pas. Afin de cadrer correctement toutes ces nouvelles possibilités diagnostiques, souvent complexes, et d'anticiper l’apparition de nouveaux tests diagnostiques, la réglementation devait être entièrement repensée.
Lors de l'élaboration d'un nouveau cadre, l'Europe a délibérément opté pour un règlement plutôt qu'une directive. Une directive peut être transposée dans la législation nationale de chaque État membre. Cela offre une certaine flexibilité. Cependant, les défis liés à la sécurité et à la qualité des dispositifs médicaux de diagnostic dépassent les frontières nationales, d’où le choix d'un règlement, qui doit être appliqué de manière quasi identique par tous les États membres.
Quelles sont les principales nouveautés par rapport à la directive IVDD ?
L'IVDR compte 160 pages, contre 37 pour l'IVDD. Les différences sont donc nombreuses. Les plus importantes concernent les exigences auxquelles doivent satisfaire les DIV et leur contrôle.
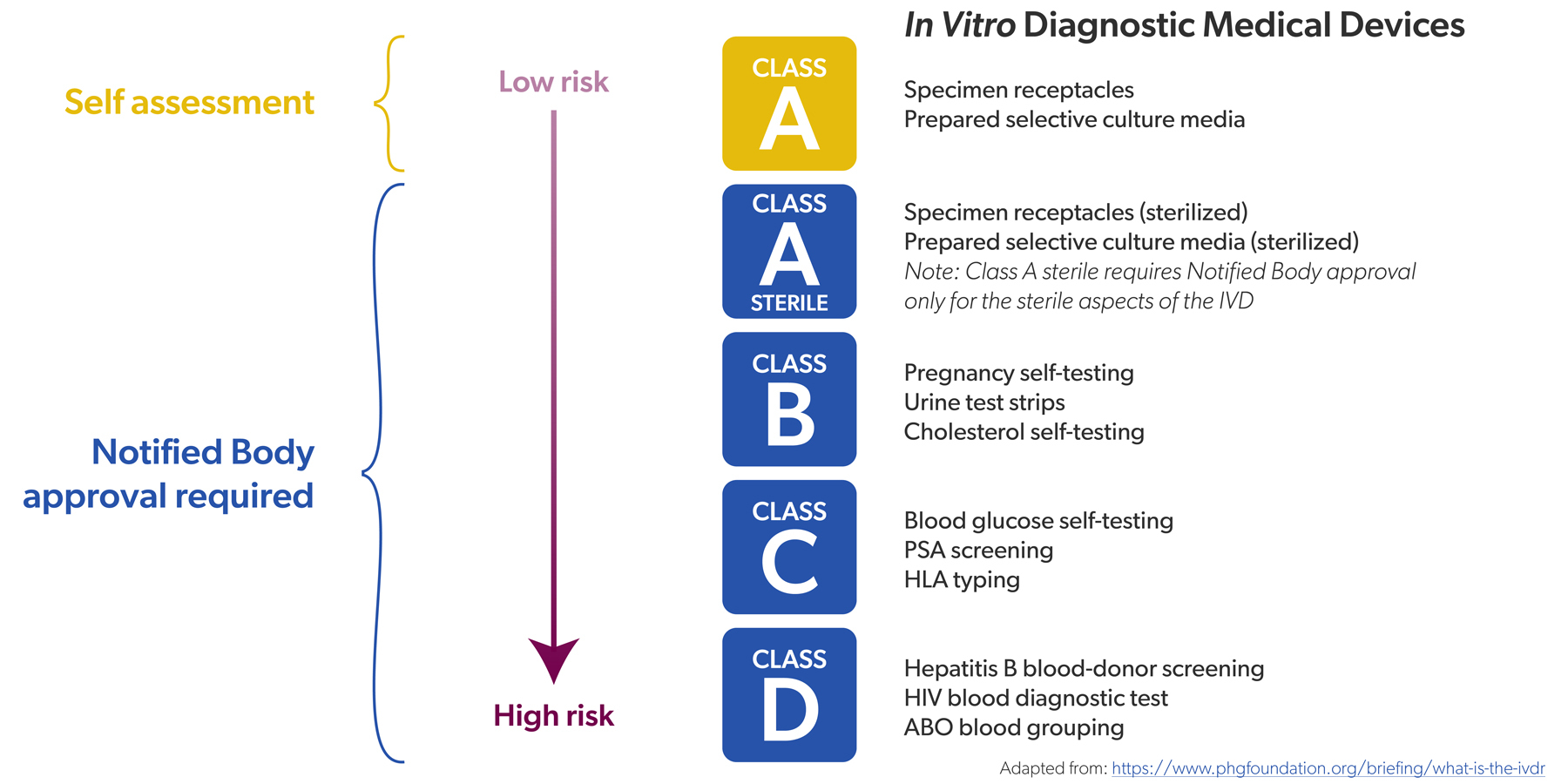
Auparavant, sous l'IVDD, un nombre limité de DIV étaient classés comme « à haut risque ». Il s'agissait par exemple des tests de dépistage du VIH ou de l'hépatite. Ces DIV devaient faire l'objet d'une évaluation de conformité par un organisme notifié – cela concernait environ 20 % de tous les DIV – avant de pouvoir être mis sur le marché. Pour les 80 % restants, une auto certification par le fabricant suffisait.
« Auparavant, 20 % de tous les DIV devaient être contrôlés par un organisme notifié, contre 80 % aujourd'hui. »
Dans le cadre de l’IVDR, tous les DIV ont reçu une classe de risque (de faible à élevée, de A à D) et le rapport s'est inversé : environ 80 % de tous les DIV doivent désormais être contrôlés par un organisme notifié, contre 20 % auparavant. Seuls les DIV non stériles de classe A peuvent encore faire l'objet d'une autocertification, par exemple les réactifs de couleur.
Les évaluations de conformité sont également plus strictes, tout comme la surveillance des organismes notifiés qui les effectuent. Pour les DIV à haut risque, il existe désormais des acteurs supplémentaires qui participent à l'évaluation de la conformité, tels que le comité d'experts en DIV et les laboratoires de référence européens. Leur expertise est un atout important.
Concrètement, qu'implique une évaluation de conformité plus stricte ?
Les exigences en matière de sécurité des produits, d'identification et de traçabilité ont été considérablement renforcées. La barre est également placée beaucoup plus haut en termes de performances. Avant qu'un DIV puisse être mis sur le marché, il faut désormais fournir beaucoup plus de preuves cliniques de l'efficacité du dispositif.
Sous l'IVDD, une déclaration du fabricant indiquant qu'une étude de performance serait réalisée suffisait. L'IVDR impose une charge de la preuve beaucoup plus importante, en particulier pour les études présentant un risque plus élevé pour les patients participants.
Toute personne souhaitant mener une telle étude doit en outre obtenir l’accord préalable d’un comité d’éthique et de l’autorité compétente.
« Un projet pilote européen visant à simplifier la procédure de demande pour les études multinationales a récemment été lancé. »
Parallèlement, l'Europe cherche des moyens de réduire la charge administrative. Dans ce contexte, un projet pilote a récemment été lancé, dans le cadre duquel les promoteurs qui souhaitent mener une étude dans plusieurs États membres pourront introduire une demande unique. L'évaluation de cette demande sera coordonnée sous la direction d'une seule autorité compétente, plutôt que par chaque État membre séparément.
En matière de charge administrative, MedTech Europe met en garde contre les difficultés que ces exigences supplémentaires font peser sur les entreprises, qui risquent de rendre certains DIV indisponibles dans l'UE. En Belgique, nous ne constatons pour l'instant aucun problème, mais nous surveillons la situation rigoureusement. Quel est le point de vue de l'AFMPS à ce sujet ?
En tant que pouvoirs publics, nous sommes bien conscients des risques potentiels et suivons la situation de près. L’UE en est également consciente. C'est pourquoi elle a par exemple prolongé les périodes de transition pour les DIV déjà commercialisés sous la directive IVDD. Afin d'avoir une meilleure vue d'ensemble des pénuries éventuelles, elle a récemment introduit l'obligation pour les fabricants de signaler à l'avance aux autorités et aux clients les pénuries prévues de dispositifs médicaux importants.
En Belgique, nous ne constatons effectivement aucun problème urgent. À l'heure actuelle, nous n'avons connaissance que d'un test de dépistage de la syphilis important, fabriqué par une entreprise asiatique, qui ne sera plus commercialisé en Europe. Mais la Belgique est avant tout un pays de distribution : les fabricants de DIV sont principalement actifs dans d'autres pays. Par conséquent, nous ne sommes généralement pas les premiers informés lorsqu'un fabricant décide d'arrêter la production d'un test.
Notre appel à beMedTech et à vos membres est donc clair : si vous sentez que quelque chose se prépare, avant que cela ne devienne un véritable problème, n'hésitez pas à nous en faire part. Nous mettrons alors tout en œuvre pour trouver des solutions ensemble.
C'est comme pour les problèmes de santé : plus on intervient tôt, plus on a de chances d'éviter une aggravation. Vos membres sont les oreilles dont nous avons besoin pour détecter les problèmes potentiels le plus tôt possible.