Actualités et publications
Grâce à notre salle de presse, nous partageons régulièrement des mises à jour sur les technologies médicales, les évolutions politiques pertinentes et des publications intéressantes. Vous restez ainsi informé d’un secteur en constante évolution.

Jeroen Poels, AFMPS: « L'IVDR doit être une histoire positive pour tout le monde » (partie 2/2)
La semaine dernière, nous avons discuté avec Jeroen Poels, expert en DIV au sein de l'AFMPS, des exigences plus strictes de l'IVDR et de leur impact administratif sur les entreprises. Aujourd'hui, nous élargissons le débat et examinons la signification de l'IVDR pour les différentes parties prenantes.
Monsieur Poels, la semaine dernière, nous avons conclu sur la charge administrative que représente l'IVDR pour les entreprises. Plus d'administration signifie également des coûts plus élevés pour ces entreprises. L’AFMPS le constate-t-il ?
La transition vers la nouvelle législation ne doit en effet pas être sous-estimée pour les entreprises du secteur des DIV. Pour les entreprises de dispositifs médicaux (MD), la réglementation a également changé avec l'arrivée du règlement sur les dispositifs médicaux (MDR), mais les modifications sont moins radicales que celles prévues par l'IVDR.
« Les entreprises du secteur des DIV sont souvent des PME qui ne disposent pas de moyens considérables. »
De plus, de nombreuses entreprises DIV sont des PME qui ne disposent pas des mêmes moyens que les fabricants MD, plus souvent de grande taille. L'Europe tente d'en tenir compte, par exemple avec le paquet de mesures en faveur des PME (2023) et une stratégie européenne spécifique pour les start-ups et les scale-ups (2025).
En reportant l'entrée en vigueur de l'IVDR et en prévoyant des périodes de transition supplémentaires, l'Europe souhaite donner à toutes les parties prenantes concernées suffisamment de temps pour se mettre en conformité.
La pression sur les entreprises du secteur des DIV reste toutefois réelle. Le renforcement des exigences en matière de preuves cliniques et les contrôles effectués par les organismes notifiés et, éventuellement, par les laboratoires de référence européens entraîneront inévitablement une augmentation des coûts pour les entreprises. Nous entendons donc que les fabricants de DIV évaluent de manière critique leur portefeuille de produits : quels produits souhaitent-ils « transférer » vers le cadre de l'IVDR et lesquels souhaitent-ils abandonner ?
Le renforcement de l'IVDR rend inévitablement l'innovation encore plus coûteuse qu'elle ne l'était déjà. Craignez-vous que l'IVDR freine l'innovation ?
C'est difficile à prédire aujourd'hui, mais j'espère que non. En tout état de cause, l'Europe ne veut pas en arriver là et a créé un groupe de travail chargé d'accélérer les procédures relatives aux « innovations de rupture ».
En tant qu'autorité très appréciée au sein de l'UE, l'AFMPS participe pleinement à cette initiative.
L'AFMPS est en effet une marque internationale forte en Europe. Cela ressort clairement du rôle très actif que vous jouez, entre autres, au sein du Medical Device Coordination Group (MDCG).
Notre administration est en effet très active, et ce dans les 13 groupes de travail du MDCG. Ces groupes de travail traitent tous les aspects pertinents pour la mise en œuvre de l'IVDR et du MDR, tels que la notification et le contrôle des organismes notifiés, la vigilance, le contrôle du marché, les études cliniques et de performance, etc.
« Les autorités compétentes d'autres États membres font régulièrement appel à l'expertise belge en matière de DIV. »
Un groupe de travail se concentre spécifiquement sur l'interprétation et la mise en œuvre homogènes des DIV , et conseille les 12 autres groupes sur des sujets spécifiques aux DIV. Depuis 2022, ce groupe DIV a publié pas moins de 16 documents d'orientation. En collaboration avec l'AFMPS, nous avons contribué à chacun de ces 16 documents, souvent en tant que chef de file. Les autorités compétentes des autres États membres de l'UE reconnaissent cette expertise et sollicitent régulièrement notre avis sur des sujets liés aux DIV.
J'ai d'ailleurs récemment été nommé coprésident du groupe de travail sur les DIV orphelins, qui sont des tests destinés à un très petit groupe de patients. Avec ce groupe de travail, nous examinons comment rendre les DIV orphelins plus facilement et plus rapidement disponibles en Europe.
Nous avons déjà longuement évoqué la pression supplémentaire que l'IVDR impose aux entreprises. Qu'en est-il de la pression sur votre administration ?
La base de données EUDAMED devrait à terme nous aider à alléger une partie de la charge administrative, mais ce n'est pas encore le cas aujourd'hui. Entre-temps, de nombreuses tâches ont été élargies et nous en avons reçu de nouvelles. Donc oui, l'IVDR entraîne également une pression supplémentaire pour nous, surtout à court terme.
Dans le même temps, il y a des signes positifs importants. On a longtemps craint, à juste titre, que la capacité des organismes notifiés ne soit pas suffisante pour l'IVDR, le nombre de DIV devant faire l'objet d'une évaluation de conformité étant passé de 20 à 80 % (voir également la première partie de l'interview). Mais ce problème semble se résoudre.
Sur les 17 organismes notifiés pour l'IVDR, seuls deux ne prennent plus de nouveaux clients, selon une enquête récente. L'avant-dernier organisme notifié à figurer sur la liste est SGS Belgium. Notre pays dispose donc désormais d'un organisme notifié pour l'IVDR, ce qui est évidemment une bonne nouvelle pour les fabricants belges !

Nous constatons également une augmentation du nombre de demandes d'études de performance. Il s'agit souvent d'études combinées avec un médicament. Le test DIV sert alors à sélectionner les patients pour l'essai et peut être commercialisé ultérieurement comme diagnostic compagnon. Un grand projet européen, COMBINE, est actuellement en cours afin de simplifier et d'harmoniser les demandes d'études combinées.
Ces signaux sont très importants. Après tout, l'IVDR doit être une réussite.
Absolument, en premier lieu pour les patients et la santé publique.
Les avantages potentiels de l'IVDR pour les patients sont évidents :Plus les tests de diagnostic in vitro sont fiables et efficaces, plus ils bénéficient au patient . Et grâce à une meilleure traçabilité, les dispositifs posant problème peuvent être localisés de manière très précise.
« Plus les tests de diagnostic in vitro sont fiables et efficaces, plus ils bénéficient au patient»
Toutes les mesures prévues par l'IVDR y contribuent plus ou moins grandement. Il est inévitable qu'un changement réglementaire d'une telle ampleur soulève de nombreuses questions. Mais celles-ci ne doivent pas remettre en cause l'ensemble de la réforme.
Il est important d'évaluer l'IVDR de manière continue et rigoureuse et d'oser l'ajuster si nécessaire : que peut-on simplifier, quelles exigences offrent trop peu de valeur ajoutée et peuvent donc être modifiées ou supprimées, etc. L'Europe recherche activement ce retour d'information. Ainsi, une consultation publique des parties prenantes sur l'IVDR et le MDR s'est achevée fin mars et ses résultats sont actuellement en cours d'analyse. Nous continuons également à recueillir les commentaires des parties prenantes concernées en Belgique par l'intermédiaire de l'AFMPS.

Jeroen Poels, AFMPS: « Les entreprises peuvent être nos antennes » (partie 1/2)
Depuis la fin du mois dernier, l'IVDR est en vigueur depuis 3 ans. Comment l'AFMPS a-t-elle vécu cette nouvelle réglementation jusqu'à présent ? Qu'est-ce qui fonctionne bien et qu'est-ce qui peut être amélioré ? Nous avons discuté avec Jeroen Poels, expert en DIV au sein de l'AFMPS. « Plus nous détectons tôt les problèmes potentiels liés à l'IVDR, plus nous pouvons agir. Mais pour cela, nous avons besoin de l'aide des entreprises. »
Le règlement européen sur les dispositifs médicaux de diagnostic in vitro, ou IVDR, est applicable depuis le 26 mai 2022. Il remplace la directive sur les dispositifs médicaux de diagnostic in vitro (IVDD). Son objectif ? Améliorer la sécurité et la qualité des dispositifs médicaux de diagnostic in vitro (DIV), renforcer la transparence et la traçabilité des DIV et harmoniser la mise en œuvre de la législation au sein de l'Union Européenne (UE)
Dans une interview en deux parties, Jeroen Poels décortique l'IVDR. Il est expert en dispositifs médicaux pour le diagnostic in vitro à l'Agence fédérale des médicaments et des produits de santé (AFMPS), qui veille à la sécurité, à la qualité et à l'efficacité des dispositifs médicaux sur le marché belge.
Monsieur Poels, commençons par le commencement. Pourquoi fallait-il remplacer la directive IVDD ?
L'IVDD datait de 1998, à une époque où les possibilités diagnostiques étaient beaucoup plus limitées qu'aujourd'hui. À l'époque, par exemple, les tests génétiques, les tests au chevet du patient (point of care tests ou POCT), les logiciels DIV, etc. n'existaient pratiquement pas. Afin de cadrer correctement toutes ces nouvelles possibilités diagnostiques, souvent complexes, et d'anticiper l’apparition de nouveaux tests diagnostiques, la réglementation devait être entièrement repensée.
Lors de l'élaboration d'un nouveau cadre, l'Europe a délibérément opté pour un règlement plutôt qu'une directive. Une directive peut être transposée dans la législation nationale de chaque État membre. Cela offre une certaine flexibilité. Cependant, les défis liés à la sécurité et à la qualité des dispositifs médicaux de diagnostic dépassent les frontières nationales, d’où le choix d'un règlement, qui doit être appliqué de manière quasi identique par tous les États membres.
Quelles sont les principales nouveautés par rapport à la directive IVDD ?
L'IVDR compte 160 pages, contre 37 pour l'IVDD. Les différences sont donc nombreuses. Les plus importantes concernent les exigences auxquelles doivent satisfaire les DIV et leur contrôle.
Auparavant, sous l'IVDD, un nombre limité de DIV étaient classés comme « à haut risque ». Il s'agissait par exemple des tests de dépistage du VIH ou de l'hépatite. Ces DIV devaient faire l'objet d'une évaluation de conformité par un organisme notifié – cela concernait environ 20 % de tous les DIV – avant de pouvoir être mis sur le marché. Pour les 80 % restants, une auto certification par le fabricant suffisait.
« Auparavant, 20 % de tous les DIV devaient être contrôlés par un organisme notifié, contre 80 % aujourd'hui. »
Dans le cadre de l’IVDR, tous les DIV ont reçu une classe de risque (de faible à élevée, de A à D) et le rapport s'est inversé : environ 80 % de tous les DIV doivent désormais être contrôlés par un organisme notifié, contre 20 % auparavant. Seuls les DIV non stériles de classe A peuvent encore faire l'objet d'une autocertification, par exemple les réactifs de couleur.
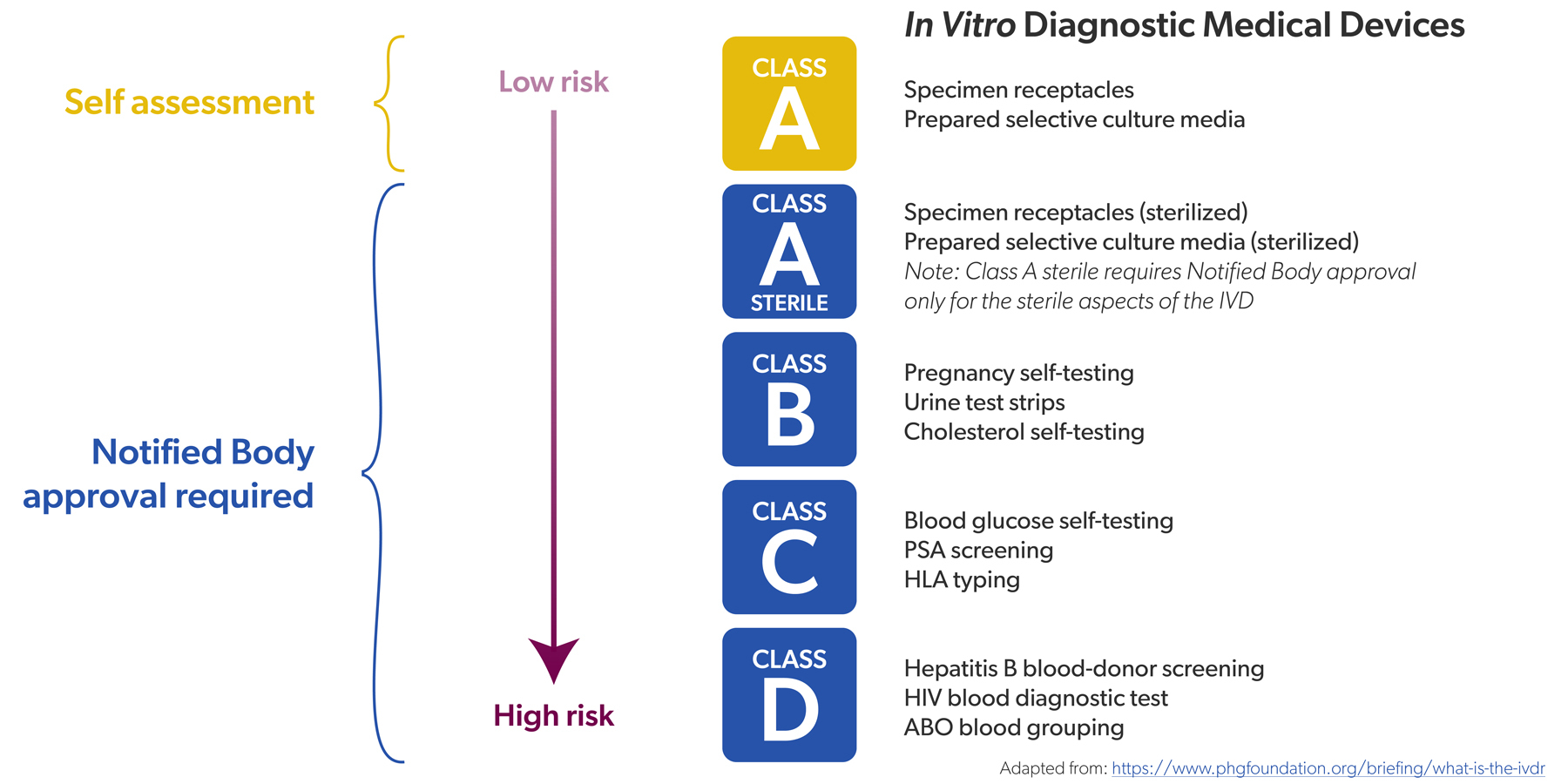
Les évaluations de conformité sont également plus strictes, tout comme la surveillance des organismes notifiés qui les effectuent. Pour les DIV à haut risque, il existe désormais des acteurs supplémentaires qui participent à l'évaluation de la conformité, tels que le comité d'experts en DIV et les laboratoires de référence européens. Leur expertise est un atout important.
Concrètement, qu'implique une évaluation de conformité plus stricte ?
Les exigences en matière de sécurité des produits, d'identification et de traçabilité ont été considérablement renforcées. La barre est également placée beaucoup plus haut en termes de performances. Avant qu'un DIV puisse être mis sur le marché, il faut désormais fournir beaucoup plus de preuves cliniques de l'efficacité du dispositif.
Sous l'IVDD, une déclaration du fabricant indiquant qu'une étude de performance serait réalisée suffisait. L'IVDR impose une charge de la preuve beaucoup plus importante, en particulier pour les études présentant un risque plus élevé pour les patients participants.
Toute personne souhaitant mener une telle étude doit en outre obtenir l’accord préalable d’un comité d’éthique et de l’autorité compétente.
« Un projet pilote européen visant à simplifier la procédure de demande pour les études multinationales a récemment été lancé. »
Parallèlement, l'Europe cherche des moyens de réduire la charge administrative. Dans ce contexte, un projet pilote a récemment été lancé, dans le cadre duquel les promoteurs qui souhaitent mener une étude dans plusieurs États membres pourront introduire une demande unique. L'évaluation de cette demande sera coordonnée sous la direction d'une seule autorité compétente, plutôt que par chaque État membre séparément.
En matière de charge administrative, MedTech Europe met en garde contre les difficultés que ces exigences supplémentaires font peser sur les entreprises, qui risquent de rendre certains DIV indisponibles dans l'UE. En Belgique, nous ne constatons pour l'instant aucun problème, mais nous surveillons la situation rigoureusement. Quel est le point de vue de l'AFMPS à ce sujet ?
En tant que pouvoirs publics, nous sommes bien conscients des risques potentiels et suivons la situation de près. L’UE en est également consciente. C'est pourquoi elle a par exemple prolongé les périodes de transition pour les DIV déjà commercialisés sous la directive IVDD. Afin d'avoir une meilleure vue d'ensemble des pénuries éventuelles, elle a récemment introduit l'obligation pour les fabricants de signaler à l'avance aux autorités et aux clients les pénuries prévues de dispositifs médicaux importants.
En Belgique, nous ne constatons effectivement aucun problème urgent. À l'heure actuelle, nous n'avons connaissance que d'un test de dépistage de la syphilis important, fabriqué par une entreprise asiatique, qui ne sera plus commercialisé en Europe. Mais la Belgique est avant tout un pays de distribution : les fabricants de DIV sont principalement actifs dans d'autres pays. Par conséquent, nous ne sommes généralement pas les premiers informés lorsqu'un fabricant décide d'arrêter la production d'un test.
Notre appel à beMedTech et à vos membres est donc clair : si vous sentez que quelque chose se prépare, avant que cela ne devienne un véritable problème, n'hésitez pas à nous en faire part. Nous mettrons alors tout en œuvre pour trouver des solutions ensemble.
C'est comme pour les problèmes de santé : plus on intervient tôt, plus on a de chances d'éviter une aggravation. Vos membres sont les oreilles dont nous avons besoin pour détecter les problèmes potentiels le plus tôt possible.

Pr Cavalier : « La médecine de laboratoire est un investissement essentiel pour des soins de qualité »
Lors d’EuroMedLab 2025, nous avons parlé avec Etienne Cavalier, professeur de chimie clinique à l’Université de Liège, chef du service de chimie clinique au CHU de Liège et président de la Royal Belgian Society of Laboratory Medicine. Quelles mesures prendrait-il en matière de médecine de laboratoire s’il était ministre fédéral de la Santé ?
« Ma priorité absolue serait de garantir un accès plus rapide aux nouveaux tests à forte valeur ajoutée. Actuellement, il faut souvent des années avant qu’un remboursement soit accordé — si tant est qu’il le soit un jour. Par exemple, les tests NT-proBNP et BNP ne sont toujours pas remboursés en Belgique, alors qu’ils figurent dans toutes les recommandations. Ce délai excessif est préjudiciable pour le patient belge. »
Retombées économiques
Comme le professeur Mario Plebani (*), le professeur Cavalier est convaincu qu’il ne s’agit pas d’un problème de budget.
(*) À l’occasion d’EuroMedLab 2025, nous avons également rencontré le professeur Mario Plebani, président de la European Federation of Clinical Chemistry and Laboratory Medicine. Lire l’interview ICI.
« Tout d’abord, de nombreux nouveaux tests permettent en réalité des économies. À court terme, en remplaçant des procédures plus coûteuses et souvent invasives. Et à plus long terme, en réduisant les besoins en soins grâce à une meilleure détection. Malheureusement, ces effets ne sont pas pris en compte dans l’évaluation du remboursement. »
« Par ailleurs, certains tests encore remboursés aujourd’hui ont une valeur clinique limitée. Et pour certains, on pourrait réduire la fréquence de prescription. Cela libérerait également un espace budgétaire pour intégrer de nouveaux tests. », poursuit le professeur Cavalier.
Expertise des professionnels du laboratoire
Mais la médecine de laboratoire ne se limite bien sûr pas aux seuls tests. L’expertise des professionnels du laboratoire est tout aussi essentielle. Si le professeur Cavalier était ministre de la Santé, il ferait de la valorisation de cette expertise sa deuxième priorité.
« Les laboratoires ont un rôle crucial à jouer dans des domaines comme la détection précoce et la stratification des risques. Mais nous assumons trop peu ce rôle de spécialiste — ou bien on ne nous en donne pas suffisamment l’occasion. »
« En tant qu’experts de laboratoire, nous devrions être associés aux discussions avec les autres spécialistes cliniques impliqués dans les parcours de soins interdisciplinaires. »
« Nous produisons parfois des résultats de tests qui, malheureusement, sont trop peu exploités par la suite. Ce sont des occasions manquées d’améliorer les soins au patient. En tant qu’experts de laboratoire, nous devrions être associés aux discussions avec les autres spécialistes cliniques impliqués dans les parcours de soins interdisciplinaires. »
« Cela exige un changement de regard sur la médecine de laboratoire. Trop d’acteurs continuent de considérer notre discipline uniquement comme un coût, alors qu’un bon diagnostic est en réalité un investissement dans des soins de qualité, centrés /fondés sur la valeur. »

« La médecine de laboratoire est restée trop longtemps une profession sans visage »
Une discipline technique, détachée des « véritables soins ». C’est ainsi que beaucoup perçoivent la médecine de laboratoire. Une vision totalement erronée, selon le professeur Mario Plebani. « La médecine de laboratoire ne peut pas être considérée comme une prestation banalisée. Nous jouons un rôle en tant qu’acteur clinique », affirme le président de la European Federation of Clinical Chemistry and Laboratory Medicine (EFLM).
À l’occasion d’EuroMedLab 2025, nous avons rencontré le professeur Mario Plebani, une autorité internationale en matière de médecine de laboratoire et président de l’EFLM. Grâce à une série de publications scientifiques, il est devenu ces dernières années l’un des principaux défenseurs de la value-based laboratory medicine.
Professeur Plebani, le concept de soins de santé fondés sur la valeur connaît un succès croissant à l’échelle internationale. Comment la médecine de laboratoire s’inscrit-elle dans cette évolution ?
La médecine de laboratoire est au cœur même des soins de santé fondés sur la value based healthcare. Pourtant, ce n’est pas encore perçu ainsi. Beaucoup — y compris parmi les décideurs politiques — considèrent notre discipline comme une activité purement technique, confinée en coulisses. Et pourtant, nous intervenons dans presque tous les parcours de soins : du dépistage au diagnostic, jusqu’au traitement et au suivi.
Comment expliquer alors que notre rôle soit encore si souvent sous-estimé ou oublié ?
Cela tient en grande partie à notre invisibilité physique. Nous travaillons dans des laboratoires, loin des soins cliniques visibles. Les patients ne nous voient pas. Même de nombreux confrères médecins ne nous voient pas réellement. Ils consultent les résultats d’analyse, sans toujours reconnaître l’expertise nécessaire pour les produire.
Vous dites parfois que vous et vos collègues devez « sortir de votre silo »…
En effet. Il faut donner un visage à la médecine de laboratoire, et cela passe nécessairement par une meilleure communication avec les médecins, les patients et les décideurs.
Notre état d’esprit doit également évoluer.
Pendant longtemps, nous avons concentré nos efforts sur le délai de réponse et le coût par test. Cela a pu donner l’impression, à tort, que nous n’étions qu’une unité de production déconnectée des soins. Bien sûr, cette approche nous a permis de gagner en efficacité. Mais l’efficacité n’est qu’une partie de l’équation.
Il est grand temps de passer à l’étape suivante : mettre davantage l’accent sur la qualité et l’impact, plutôt que sur le volume et le coût. Voilà l’essence même de la based laboratory medicine.
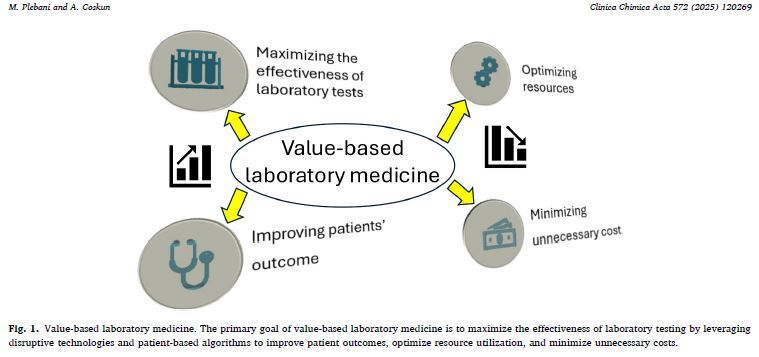
Image tirée de 'Promoting value-based laboratory medicine: Moving towards an innovative model of clinical laboratory''Promoting value-based laboratory medicine: Moving towards an innovative model of clinical laboratory' (Plebani, M., & Coskun, A., 2025)
Quelle est votre responsabilité, en tant que secteur, dans cette transformation ?
Cela commence par une prise de conscience plus forte de notre propre rôle. Oui, une part de notre travail est technique. Mais notre responsabilité ne s’arrête pas à la livraison de résultats bruts. Il s’agit de transformer les données en informations cliniquement pertinentes pour les équipes soignantes, de les intégrer dans la prise de décision, d’accompagner les parcours patients avec des données longitudinales, de créer des liens avec d’autres disciplines…
Prenez l’exemple des patients atteints d’insuffisance cardiaque. Les dosages sanguins des peptides natriurétiques (tels que NT-proBNP et proBNP, ndlr) pourraient permettre d’éviter un grand nombre d'investigations supplémentaires moins utiles. Mais leur interprétation requiert une expertise particulière.
Cela signifie que nous devons pleinement assumer un rôle clinique dans ce parcours de soins, au-delà d’un simple appui en arrière-plan.
Certains craignent toutefois qu’un rôle renforcé de la médecine de laboratoire n’entraîne une hausse du nombre de tests — et donc des coûts. Que répondez-vous à cela ?
Je comprends cette crainte, mais elle n’est pas fondée.
Il ne s’agit pas d’augmenter les budgets de la santé, mais de les utiliser autrement.
Actuellement, le financement des soins repose presque partout sur une logique de soins curatifs, plutôt que de prévention. On se concentre sur la fin du parcours, sur le traitement des maladies. Le début du continuum — comprendre comment les maladies apparaissent et comment les prévenir ou en retarder l’évolution — reste trop négligé.
Et pourtant, l’offre de soins actuelle ne peut pas absorber le flux constant de nouveaux patients nécessitant des traitements coûteux. Attention, il ne s’agit pas d’opposer curatif et préventif. Les soins curatifs restent essentiels. Mais nous devons intervenir plus tôt dans le processus.
Quel rôle la médecine de laboratoire peut-elle jouer dans ce cadre ?
Un rôle multiple. Nous pouvons contribuer à réduire l’afflux de nouveaux patients grâce au diagnostic préventif, détecter les maladies plus précocement grâce au dépistage, et améliorer le ciblage des traitements personnalisés grâce aux tests compagnons, entre autres.
Tout cela nécessite évidemment des ressources, mais ce n’est pas un « coût » : c’est un investissement. Selon moi, investir dans la value-based laboratory medicine est indispensable à la durabilité des systèmes de santé.
Le vrai obstacle aujourd’hui est que nous avons encore du mal à rémunérer la qualité plutôt que le volume. Trop de laboratoires sont encore « récompensés » en fonction du nombre de tests réalisés, indépendamment de leur pertinence.
Tant que ce modèle perdure, il sera difficile de généraliser la value-based laboratory medicine.
Que faut-il faire pour sortir de cette impasse ?
Une des clés majeures est une meilleure utilisation des données.
Nous collectons d’énormes quantités de données dans les soins de santé. Ces données sont cruciales pour la prise en charge des patients, la recherche scientifique… mais aussi pour orienter les politiques de santé, notamment en matière de financement.
Si nous voulons positionner la médecine de laboratoire comme levier stratégique pour le value based healthcare, il faut lier systématiquement les résultats de tests à des indicateurs de valeur. Un test réduit-il le risque de complications ? Diminue-t-il le nombre de réhospitalisations ? En collectant et croisant ces informations, nous pouvons documenter l’impact réel des tests et alimenter des décisions politiques fondées.
Les initiatives visant à mieux exploiter les données de santé vont donc de pair avec la transition vers une médecine de laboratoire axée sur la valeur ?
Les avancées fulgurantes dans le domaine de l’intelligence artificielle et des données de santé créent en tout cas un véritable élan pour la value-based laboratory medicine , et plus largement pour une approche value based heatlhcare. C’est indéniable. Sans ces technologies, il serait extrêmement difficile de collecter et de traiter les données à la vitesse nécessaire.
Je citerais volontiers l'exemple des valeurs de référence.
Les résultats d’analyse sont souvent interprétés en les comparant à des valeurs de référence établies sur une population générale — autrement dit, comment les résultats d’un patient donné se situent-ils par rapport à la moyenne. Il est toutefois bien plus pertinent, sur le plan clinique, de comparer les valeurs d’un patient à ses propres valeurs antérieures. Cela permet d’obtenir une image beaucoup plus précise et cliniquement exploitable. Mais pour pouvoir réaliser ce type d’analyse, il faut disposer de mesures répétées, fiables et de qualité pour ce patient, ainsi que d’un logiciel capable de traiter ce volume de données de manière robuste.
D’autres évolutions technologiques soutiennent également la transition vers une médecine axée sur la valeur.
Je pense notamment aux tests de diagnostic point-of-care, qui sont réalisés au chevet du patient, en dehors du laboratoire. Leur accessibilité est bien plus grande et ils permettent d’obtenir des résultats rapidement, ce qui aide le clinicien à orienter plus vite le patient vers le parcours de soins approprié.
Ce type de tests décentralisés constitue un complément précieux aux analyses centralisées, à condition bien entendu que leur qualité soit garantie — un impératif qui vaut pour tout type de test.
Est-ce donc simplement une question de temps avant que les systèmes de soins fondés sur la valeur s’imposent ?
J’aimerais pouvoir répondre « oui » à cette question… mais il faut rester nuancé (sourit).
La technologie seule ne suffira pas. Ce dont nous avons le plus besoin, c’est d’un large soutien, de la part des décideurs politiques mais aussi de tous les acteurs de la santé. Il faut prendre conscience qu’évoluer vers de la value-based laboratory medicine, et plus largement vers de la value-based healthcare, n’est pas une option. C’est une nécessité si nous voulons garantir à nos enfants et petits-enfants l’accès à des soins de qualité, abordables.
Quel message souhaitez-vous adresser en conclusion aux décideurs politiques ?
Avant tout, prenez conscience que la médecine de laboratoire n’est pas un service technique, mais une véritable discipline clinique. Un rôle qui ne peut plus être dissocié de la pathologie et de la radiologie, les deux autres piliers du diagnostic. Pour une prise en charge optimale des patients, il est essentiel de sortir des silos et de constituer des équipes diagnostiques intégrées.
Et enfin — c’est fondamental — associez-nous à l’élaboration des politiques de santé. Nous avons les données et l’expertise pour accélérer la transition vers des soins fondés sur la valeur. Mais nous ne pourrons jouer ce rôle qu’en étant présents autour de la table.

L’humain et non la technologie détermine la percée de l’IA
Les progrès dans le domaine de l’intelligence artificielle (IA) font de plus en plus souvent les gros titres. Les applications potentielles ou proposées frappent l’imaginaire, mais comme pour les (autres) percées scientifiques, il n’est pas toujours évident pour les personnes extérieures de savoir si elles seront déployées dans la pratique et quand elles le seront.
Certains dessinent parfois aussi des images apocalyptiques d’une société future gérée par les robots et les machines. Dans le secteur des soins de santé, on craint plus particulièrement que le contact personnel entre le prestataire de soins et le patient disparaisse et que les soins se « déshumanisent ». La réalité tend pourtant à prouver le contraire, mais les exemples positifs retiennent plus difficilement l’attention…
Des exemples positifs de l'utilisation de l'IA attirent moins l'attention que les exemples négatifs.
Dans notre secteur, l’IA est aujourd’hui plutôt utilisée pour offrir un (meilleur) soutien aux prestataires de soins et aux patients.
Certaines applications de l’IA déchargent ainsi les professionnels de la santé de leurs tâches répétitives et/ou administratives, ce qui leur permet de consacrer plus de temps au patient. D’autres exploitent les données pour mieux adapter les soins aux besoins, aux souhaits et à la qualité de vie du patient. Plus encore qu’aujourd’hui, les soins seront de plus en plus orientés vers le « sur mesure » dans les années à venir.
Prévention accrue et de meilleure qualité
Outre ce soutien direct aux prestataires de soins et aux patients, l’IA crée également des perspectives au niveau de la population.
Nous pouvons utiliser l’intelligence artificielle pour extraire de nouvelles informations de la montagne toujours croissante de données de santé et autres données pertinentes. Des informations qui nous permettront de réaliser des progrès considérables en termes de prévention (thème largement inexploré en Belgique à ce jour), de détection (précoce), de diagnostic et de traitement.
Pourrions-nous également réaliser ces avancées sans l’aide de l’IA ? C’est probable, mais cela exigerait dans tous les cas beaucoup de temps et de capacités.
D'autre part, quiconque ayant déjà essayé ChatGPT ou d’autres applications de l’IA sait qu’on ne peut pas faire aveuglément confiance à tout ce que l’IA « produit ». Pour éviter les accidents, il importe que nous en soyons tous conscients et, dans la mesure du possible, que nous comprenions (dans les grandes lignes) ce qu’il se passe sous le capot des applications d’IA.
Pour éviter les accidents, il importe que nous comprenions plus ou moins ce qu’il se passe sous le capot des applications d’IA.
Ceci n’est toutefois pas suffisant. Nous devons également organiser la « protection » au niveau de la société, par exemple en instaurant un minimum de règles pour garantir une application correcte et sûre de l’IA.
AI Act européen

Dans ce contexte, l’Union européenne a récemment annoncé la mise en place du tout premier cadre législatif de grande envergure, à travers la loi européenne sur l’intelligence artificielle ou « EU AI Act ».
Grâce à cette législation, les applications problématiques de l’IA (p. ex. le « social scoring ») seront interdites, la commercialisation des applications à haut risque (p. ex. applications médicales d’IA) sera soumise à des exigences précises et il sera obligatoire d’indiquer où et quand l’IA est utilisée (p. ex. dans le cas d’avatars ou de chatbots alimentés par l’IA).
Dans le secteur des soins de santé, une législation stricte est déjà en place depuis un certain temps en ce qui concerne les technologies médicales, y compris celles qui sont pilotées par des logiciels et par l’IA. L’EU AI Act va plus loin et prévoit des garanties supplémentaires, notamment en matière de transparence, de surveillance et d’évaluation. Un moyen d’ancrer plus solidement les principes éthiques dans la législation.
Au risque de vous décevoir, ceci n’ouvrira pas immédiatement la voie à un déploiement à grande échelle des applications d’IA dans les soins de santé. Beaucoup de questions doivent en effet encore être clarifiées.
De nouvelles questions
L’utilisation de l’IA dans les soins de santé soulève donc de nouvelles questions en ce qui concerne la démontrabilité et la mesurabilité des modèles d’IA (pouvez-vous expliquer ce qu’ils font, pourquoi et comment ?), des aspects cruciaux lorsqu'il s'agit du financement/remboursement des applications.
Plus que jamais, l'aspect humain sera déterminant dans la percée de l’IA.
Il convient de renforcer les connaissances et la confiance dans l’IA, tant dans le chef des prestataires de soins que dans celui des patients. Le professionnel de la santé de demain devra absolument être capable de travailler avec les technologies de l’IA. Il devra être en mesure d’évaluer si une certaine application d’IA peut compléter ses connaissances et son expertise, de sorte à prendre de meilleures décisions et à comprendre les limites et les risques qui vont de pair.
Une communication claire et transparente entre le prestataire de soins et le patient restera la pierre angulaire de soins axés sur la personne, y compris à l’avenir.
Une communication transparente avec le patient restera dans tous les cas la pierre angulaire de soins axés sur la personne. Il incombe au prestataire de soins et au patient d’évaluer, en concertation, si certaines technologies d’IA peuvent être intégrées dans le programme de soins et, si oui, lesquelles, étant entendu qu’elles doivent l’être au profit de la santé, du bien-être et de la qualité de vie du patient.
Au-delà du battage médiatique
Si les médias prédisent presque quotidiennement des scénarios utopiques ou dystopiques sur « l’avenir avec l’IA », à nous de porter notre regard au-delà du battage médiatique. Comme pour chaque nouvelle technologie, nous devons envisager l’IA de manière critique et réaliste. L’IA offre certes de formidables opportunités, mais il n’est jamais bon d’avancer tête baissée.
Acquérir pas à pas de nouvelles connaissances sur l’IA dans le secteur des soins de santé et introduire progressivement de nouvelles applications d’IA dans la pratique, voilà la seule manière d’instaurer durablement la confiance dans le chef des patients, des prestataires de soins et des décideurs politiques. Tout le monde parle (à juste titre) de la « médecine fondée sur les faits ». Respectons cela pour l’IA également, en sachant que la collecte de données probantes prend du temps.
Au final, les soins de santé ne dépendent pas tant de l’une ou l’autre technologie, mais plutôt de la manière dont nous, en tant que société, pouvons organiser au mieux les soins et les proposer à tous.
À propos de l'auteur
Danny Van Rojen est un expert politique spécialisé dans la politique européenne en matière de technologie numérique, et plus largement de politique de santé.

Il a contribué à différentes publications internationales sur l’utilisation et les applications éthiques dans les soins de santé et a donné plusieurs conférences à ce sujet, notamment pour la Commission européenne et l’Agence européenne des médicaments. Il a également été membre du groupe de parties prenantes eHealth de la Commission européenne.
Contact? LinkedIn

Nouvelle mise à jour de la charte IVD souligne l’engagement du secteur.
La charte beMedTech pour le diagnostic in vitro fait peau neuve. La charte a été mise à jour à la suite de plusieurs modifications de la réglementation DIV. Les Commissions de Biologie Clinique et d’Anatomie Pathologique ainsi que le Collège belge pour l’Hérédité Humaine et les Maladies Rares approuvent cette mise à jour.
Cette nouvelle mise à jour intervient un an et demi après la précédente, réalisée fin 2023, et tient compte des évolutions de la réglementation européenne depuis lors.
Par exemple, la période de transition du Règlement IVDR a été prolongée, permettant ainsi à toutes les parties prenantes de disposer de plus de temps pour certifier les dispositifs médicaux de diagnostic in vitro conformément à la réglementation.
« Garantir qualité et sécurité »
« En 2016, nous avons publié notre toute première charte DIV en réponse aux demandes des laboratoires médicaux. En 2023, une révision approfondie s’imposait, et aujourd’hui, une deuxième mise à jour s’en suit », explique Lieselot De Vos, conseillère en DIV auprès de notre fédération.
« Cela montre que nous restons à jour en tant que secteur, ce qui est absolument essentiel », ajoute Lieselot De Vos. « Les dispositifs de diagnostic in vitro jouent un rôle clé dans le processus de prise de décision des médecins. Il est crucial que tous les acteurs impliqués puissent compter sur du matériel sûr et de haute qualité. »
Les Commissions de Biologie Clinique et d’Anatomie Pathologique ainsi que le Collège belge pour l’Hérédité Humaine et les Maladies Rares ont validé cette nouvelle version.
10 engagements concrets
Les 23 membres DIV de beMedTech ont tous signé la charte mise à jour. Les entreprises s’engagent à soutenir les laboratoires médicaux dans le respect des exigences réglementaires et dans leurs efforts de qualité.
Elles le font à travers dix engagements concrets, entre autres le respect strict des lois et réglementations, la formation adéquate du personnel technique, la protection des données personnelles et un accent mis sur la cybersécurité.

L'IA au service de la radiothérapie à la Clinique Saint-Jean
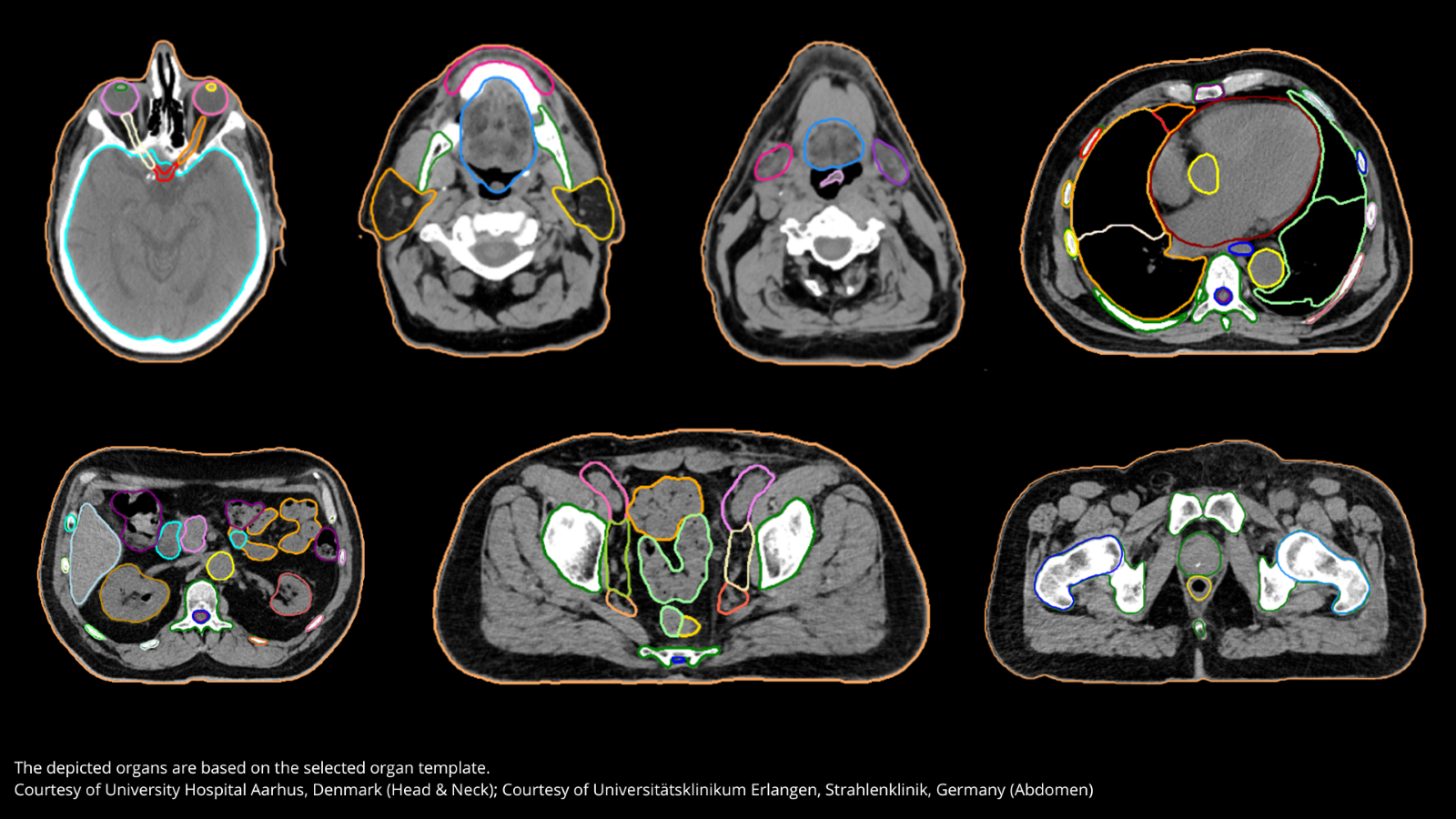
Dans le traitement du cancer, la radiothérapie joue un rôle crucial. Cette thérapie utilise des rayonnements de haute énergie pour détruire les cellules cancéreuses. Dans ce contexte, la précision est cruciale, notamment lorsqu'il s'agit de contourer les organes à risque pour éviter des dommages collatéraux. Bien que nécessaire, cette étape peut être chronophage et complexe. Heureusement, des outils innovants peuvent accélérer ce processus, explique le Dr Sophie Cvilic (Clinique Saint-Jean).
La Clinique Saint-Jean à Bruxelles a récemment adopté l’AI-Rad Companion Organs RT, une technologie basée sur l'intelligence artificielle pour automatiser le contourage des organes à risque et des volumes tumoraux. Le Dr Sophie Cvilic (photo) chef du service de radiothérapie, témoigne des avancées.

« Avant, mes technologues en imagerie médicale passaient plus d'une heure à dessiner manuellement ces organes. Désormais, ce travail est effectué en 10 à 20 minutes avec une précision impressionnante. » La solution permet en outre de délimiter des structures complexes comme le plexus brachial ou les vaisseaux médiastinaux avec une qualité rarement atteinte auparavant.
« Auparavant, mes techniciens passaient plus d'une heure à délimiter les organes à risque. Maintenant, cela peut se faire en 10 à 20 minutes. »
Helena Trindade, technologue à la Clinique Saint-Jean, ajoute : « Le temps gagné est significatif. Bien que certains ajustements mineurs soient encore nécessaires, l’outil simplifie grandement notre travail quotidien et améliore l'efficacité globale du processus. »
Perfectible
Quels sont ces ajustements? Dr. Cvilic : « Certaines zones ganglionnaires, comme les zones 3 et 4, nécessitent encore une validation et des corrections manuelles. La qualité des dessins automatiques est excellente, mais il est important de rester vigilant pour s'assurer qu'aucune erreur ne se produise. »
Cela met en avant la complémentarité entre l’outil et les experts humains, soulignant que l’intelligence artificielle n’a pas pour vocation de remplacer les technologues mais bien de leur offrir un support précieux.

De son côté, Helena Trindade (photo) note que certaines structures plus petites ou spécifiques demandent encore une intervention humaine. Cela illustre l'importance du rôle des technologues dans la vérification et l'ajustement des contours, garantissant ainsi une qualité optimale.
Pourquoi l'adoption reste encore limitée
Malgré leur grand potentiel, des solutions comme celles de la Clinique Saint-Jean ne sont pas encore largement utilisées dans les centres de radiothérapie en Belgique. Selon Olivier Adant de Siemens Healthineers, cela est dû en partie au coût d'investissement initial pour les hôpitaux, au manque de financement spécifique, aux défis d'intégration des technologies dans les infrastructures existantes et à une certaine réticence à l'innovation.
« En tant que fournisseur, nous observons que ces obstacles nécessitent une approche coordonnée », explique-t-il. « Il faut d'abord résoudre la question du financement. Ensuite, il est important de prévoir un cadre approprié, notamment sous forme de formations sur le terrain. Cela est indesipensable pour une adoption fluide et garantir que les avantages de ces technologies soient réellement exploités. »
« Des normes standardisées pour l'évaluation de ces outils pourraient accélérer leur diffusion à grande échelle. »
Olivier Adant plaide enfin pour la mise en place de standards harmonisés pour évaluer et comparer les performances de ces solutions . « Cela pourrait accélérer la diffusion à plus grande échelle. »
Des pionniers
Avec l'arrivée des outils d'IA, un nouveau chapitre s'est clairement ouvert dans l'évolution de la radiothérapie. Le temps de préparation plus court et la plus grande précision permettent des soins plus personnalisés et plus efficaces.
Toutefois, leur généralisation nécessitera des efforts collectifs pour surmonter les barrières financières, techniques et culturelles. Mais des histoires comme celle de la Clinique Saint-Jean montrent que ceux qui le souhaitent peuvent déjà utiliser la technologie pour faire la différence pour les patients.

Sept apps remboursées pour la télésurveillance en cas d'insuffisance cardiaque chronique
Depuis le 1er janvier 2025, les hôpitaux reçoivent un financement du gouvernement pour la télésurveillance des patients atteints d’insuffisance cardiaque chronique. Six hôpitaux ont déjà conclu un accord à ce sujet avec l'Inami.
Frank Vandenbroucke, ministre de la Santé publique : « Nous sommes convaincus que la télésurveillance aura un impact positif sur la qualité de vie des patients atteints de problèmes cardiaques chroniques et sur celle de leur famille. Cette technique permet d’identifier et de répondre plus rapidement aux besoins du patient en matière de soins, ce qui réduira finalement le nombre de réadmissions à l’hôpital. »
La télésurveillance est utilisée depuis des années déjà pour assurer le suivi des patients à distance, mais les coûts doivent généralement être pris en charge par l’hôpital et/ou le patient. Une contrainte financière qui fait (ou plutôt faisait ?) obstacle à une véritable percée de la télésurveillance en Belgique.
Équipe de télésurveillance
La situation a toutefois récemment évolué pour les patients atteints d’insuffisance cardiaque chronique. Depuis le 1er janvier 2025, l'Inami rembourse leur suivi par télésurveillance aux hôpitaux.
Les hôpitaux ne bénéficient toutefois pas automatiquement de cette intervention. Ils doivent adhérer à la nouvelle convention « télésurveillance et orientation thérapeutique en cas d’insuffisance cardiaque chronique », qui s’accompagne de certaines conditions. L’hôpital doit, par exemple, disposer d’une équipe de télésurveillance composée au minimum d’un cardiologue et d’un infirmier spécialisé en insuffisance cardiaque. Les patients sont ainsi assurés de bénéficier d’un suivi sûr et de qualité.

« Grâce à la convention, nous pouvons suivre nos patients à domicile et ainsi réduire le nombre d’hospitalisations. »
Dr Matthias Dupont, ZOL
Jusqu'à présent, six hôpitaux ont déjà adhéré à l'accord : Ziekenhuis Oost-Limburg (Genk), Centre Hospitalier de Mouscron, AZ Sint-Maarten (Malines), VITAZ (Saint-Nicolas), AZ Klina et AZ Groeninge (Courtrai). L'Inami tient à jour sur son site web une liste de tous les hôpitaux ayant rejoint l'accord.
Le Dr Matthias Dupont (photo), chef du service de soins intensifs cardiaques au ZOL et ancien président du groupe de travail belge sur l’insuffisance cardiaque, se réjouit de cette nouvelle convention : « Il s’agit d’un grand pas en avant pour la prise en charge des personnes atteintes d’insuffisance cardiaque. Grâce à la nouvelle convention, nous pouvons désormais suivre nos patients à domicile, et ainsi réduire le nombre d’admissions à l’hôpital. »
Sept apps (dont six belges)
Les hôpitaux peuvent définir eux-mêmes la ou les technologies médicales qu’ils souhaitent utiliser pour le suivi des patients atteints d’insuffisance cardiaque. À condition toutefois que l’application choisie respecte l’ensemble des exigences décrites dans la convention de l'Inami.
Au moins sept applications sont conformes à ce jour : FibriCheck, Remecare, moveUp, Healthentia, Comunicare et Well@Home (développées en Belgique) et Comarch (développée en Pologne). Un aperçu de ces applications est disponible sur le portail mHealthBelgium.
Ministre Vandenbroucke : « Le fait qu’un si grand nombre d’applications puissent déjà être utilisées dans le cadre de cette convention est une très bonne chose. Nous sommes impatients d’avoir les premiers retours des patients. »
Autres maladies chroniques
Steven Vandeput, conseiller en santé numérique pour beMedTech, a été étroitement impliqué dans l’élaboration de la nouvelle convention sur l’insuffisance cardiaque. Il applaudit cet ancrage structurel de la télésurveillance dans le trajet de soins de l’insuffisance cardiaque chronique.
« Et ce n’est qu’un début », se réjouit-il. « La télésurveillance recèle également de nombreuses possibilités pour le suivi de problèmes cardiaques autres que l’insuffisance cardiaque chronique. À terme, nous pourrons étendre la télésurveillance à d’autres groupes de patients, comme les personnes atteintes de diabète, de cancer, d’apnée du sommeil, etc. ».
Le Dr Paul Dendale, chef du service de cardiologie à l’hôpital Jessa de Hasselt (qui adhérera aussi prochainement à la convention de l'Inami), va encore plus loin. Il s’attend, en effet, à une révolution des soins de santé dans les années à venir.
« Tout ce qui pourra être réalisé par voie numérique en termes de soins devra l’être. »
Dr Paul Dendale, Jessa Ziekenhuis
« Effectuer des opérations bancaires, planifier des voyages ou faire des achats en ligne sonne aujourd’hui comme une évidence. Cette évolution touche également le secteur des soins de santé. Tout ce qui pourra être réalisé par voie numérique en termes de soins devra l’être. Nous n’en sommes clairement pas encore là, mais cette nouvelle convention marque un jalon important. »
28,7 millions € de dépenses en moins par an
La décision de rembourser d’abord la télésurveillance pour l’insuffisance cardiaque n’est pas le fruit du hasard. On estime que 2 à 3 % de la population belge souffrent d’insuffisance cardiaque chronique, ce qui représente entre 250 000 et 350 000 personnes. L’insuffisance cardiaque pèse lourdement sur le budget des soins de santé, avec près de 300 millions d’euros de dépenses annuelles.
Steven Vandeput (beMedTech) : « La télésurveillance est bénéfique à deux niveaux : la dimension continue du suivi améliore la qualité des soins et la qualité de vie du patient, tandis que notre système de soins gagne en efficacité et en efficience. »
« Les Cliniques de l’Europe ont suivi des patients atteints d’insuffisance cardiaque par télésurveillance pendant quatre ans et ont calculé combien l’État dépenserait en moins si nous procédions ainsi pour tous les patients atteints d’insuffisance cardiaque en Belgique : l’économie serait de l’ordre de 28,7 millions d’euros par an. Des fonds qui permettraient d’aider bien d’autres patients. »

« Les companion diagnostics n’en sont qu’à leurs débuts »
Les companion diagnostics, ou biomarqueurs prédictifs, sont essentiels à la personnalisation des soins. En quoi consistent exactement ces tests diagnostiques ? Pourquoi sont-ils si importants ? Où en est la Belgique dans ce domaine ? Que nous réserve l’avenir ? Nous avons posé toutes ces questions à Anouk Waeytens, conseillère du ministre fédéral démissionnaire de la Santé publique Frank Vandenbroucke. « C’est une bonne chose que la Belgique soit considérée comme un bon exemple en matière de companion diagnostics, mais nous devons faire en sorte qu’elle le reste. »
Anouk Waeytens a été experte en médicaments pour l’Inami pendant de nombreuses années. Cette fonction l’a amenée à se concentrer sur les thérapies personnalisées. Depuis fin 2020, elle est conseillère du ministre fédéral des Affaires sociales et de la Santé publique, Frank Vandenbroucke. À ce titre, elle a participé et participe toujours activement aux différentes initiatives mises en place dans notre pays en matière de companion diagnostics.
Madame Waeytens, pouvez-vous nous expliquer brièvement ce que sont les companion diagnostics (CDx) ?
« Les CDx sont des tests médicaux qui permettent d’évaluer à l’avance l’efficacité d’un médicament chez un patient ou le degré de tolérance du patient à ce médicament. Ces tests se basent sur un ou plusieurs biomarqueurs, soit des caractéristiques qui permettent de prédire l’efficacité d’une thérapie donnée. Si le test montre que vous présentez la bonne caractéristique, le médecin peut entamer le traitement. Dans le cas contraire, il cherche une alternative. »
« Prenons l’exemple du biomarqueur BRAF V600. Il s’agit d’une mutation du gène BRAF, que nous possédons tous. Cette mutation provoque une division cellulaire incontrôlée et est souvent à l’origine du mélanome et du cancer du poumon, entre autres. Il existe des médicaments qui inhibent cette division cellulaire, mais ils ne sont utiles que si le cancer est causé par la mutation BRAF V600. Un test CDx permet de le déterminer à l’avance. »
Quel est le gros avantage ?
« Le principal avantage réside dans le gain de temps pour le patient. Le test réduit le risque que vous entamiez un traitement qui ne fonctionnera pas chez le patient et qui aura, de surcroît, des effets secondaires prévisibles. Ce n’est pas un hasard si les CDx sont jusqu’à présent essentiellement mis en place pour des maladies dans le cadre desquelles la précision et la contrainte de temps jouent un rôle majeur, comme le cancer. »
« L’utilisation des companion diagnostics réduit le risque que vous entamiez un traitement qui ne fonctionnera pas chez le patient ou qui aura des effets secondaires prévisibles. »
« Les tests sont aussi les bienvenus pour l’assurance maladie : nous dépensons moins d’argent du contribuable dans des traitements qui, au final, n’ont aucun effet sur un patient. Autrement dit, les tests permettent d’investir le plus efficacement possible dans la santé des patients. »
Les CDx sont souvent associés au cancer. Qu’en est-il des autres pathologies ?
« Non pas que le cancer soit “plus important” que d’autres maladies, mais il y a dans ce domaine un énorme besoin médical non rencontré : il n’existe pas encore d’option thérapeutique optimale pour de nombreux cancers. Quand il y en a une, la nécessité de chercher une thérapie ciblée est moindre. Autre facteur qui entre en ligne de compte : on en a longtemps su davantage sur le cheminement des cancers que sur celui d’autres maladies, ce qui a permis de mettre au point des traitements personnalisés. »
« Aujourd’hui, la recherche de médicaments personnalisés pour les maladies hormonales, cardiovasculaires, neurodégénératives, immunitaires, etc. s’intensifie. »
Les procédures de remboursement des tests CDx en Belgique n’ont, pendant longtemps, pas été alignées sur celles des traitements personnalisés. Conséquence : le test prédictif risquait d’être remboursé plus tard que la thérapie ciblée. Vous avez tenté de résoudre ce problème au cours de la dernière législature. Où en est-on aujourd’hui ?
« Nous avons lié les procédures de remboursement des thérapies ciblées et des companion diagnostics moléculaires en juillet 2019, afin que le remboursement ait lieu en même temps. Pourquoi se concentrer sur les marqueurs moléculaires ? Parce que c’est là que le besoin était le plus criant à l’époque : de nombreux marqueurs moléculaires étaient remboursés tardivement ou n’étaient pas remboursés. »
« En 2019, nous avons synchronisé le remboursement de 29 substances actives (une substance peut être remboursée pour plusieurs indications) et celui de leurs marqueurs moléculaires. Aujourd’hui, soit cinq ans plus tard, 16 nouvelles substances actives sont déjà venues s’ajouter à la liste. Grâce à ces procédures de remboursement couplées, la thérapie et le marqueur moléculaire sont désormais toujours remboursés en même temps. C’est un énorme progrès ! »
« À partir de 2026, les médicaments pourront être remboursés chez nous dès qu’ils auront reçu l’autorisation de l’EMA. Cela s'appliquere également aux médicaments personnalisés et à leurs biomarqueurs. »
« Faciliter l’accès aux traitements prometteurs était et reste l’une des principales ambitions de notre ministre. En d’autres termes, nous voulons que les patients aient toujours accès aux meilleurs traitements, surtout en cas d’urgence médicale. C’est un chantier sans fin, mais nous avons fait des progrès considérables ces dernières années. »
« Nous avons jeté les bases juridiques d’une nouvelle procédure de remboursement rapide en mai 2024. Les arrêtés d’exécution et le back-office de cette procédure sont en cours d’élaboration. Il s’agit de permettre le remboursement des médicaments dès l’obtention de l’autorisation de l’Agence européenne des médicaments (EMA) à partir du 1er janvier 2026. Grâce à la procédure couplée, ce remboursement rapide pourra également s’appliquer aux médicaments personnalisés et à leurs biomarqueurs. »
« Moins de bruit grâce à l’intelligence artificielle »
L’évolution considérable de l’intelligence artificielle (IA) peut donner et donnera un énorme coup de pouce aux companion diagnostics et aux soins personnalisés au sens large, estime Anouk Waeytens.
« L’IA peut apporter une contribution considérable à la phase R&D de nouveaux tests. De très nombreuses molécules peuvent jouer un rôle dans le cheminement d’une maladie. Trouver les bonnes molécules est un travail de Titan. L’IA peut faciliter la présélection de certains panels et donc permettre de cibler davantage les recherches. Autrement dit : de réduire considérablement le bruit. »
D’après Anouk, l’IA peut aussi intervenir dans l’interprétation des résultats des tests. « L’IA permet de rassembler une foule de données : données cliniques, données sur le mode de vie, données d’imagerie, données anatomopathologiques… Grâce aux liens qu’on peut établir de la sorte, il devient possible de déterminer beaucoup plus précisément quel traitement convient à quel type de patient. »
« Sera-ce simple ? Bien sûr que non. Mais le gouvernement ne doit pas rater le coche. »
En marge de cette procédure de remboursement couplée, vous avez aussi lancé, en 2019, un projet pilote de remboursement des tests de biologie moléculaire par séquençage de nouvelle génération. Quelle importance revêtent-ils dans le cadre des traitements personnalisés ?
« Dans certaines pathologies, il y a un seul biomarqueur pertinent, qu’il est possible de détecter à l’aide d’un test individuel. Dans d’autres maladies, plusieurs marqueurs interviennent. Vous pouvez effectuer un test diagnostique individuel pour chacun de ces marqueurs ou examiner plusieurs marqueurs en même temps au moyen d’un panel : un test de séquençage de nouvelle génération (NGS). »
« Ce genre de test est intéressant pour plusieurs raisons : vous ne devez effectuer qu’un seul test, vous ne prélevez qu’un seul échantillon chez le patient et vous gagnez du temps. Ce test peut, en outre, s’avérer financièrement plus avantageux que si vous deviez réaliser plusieurs tests individuels. »
« En 2019, nous avons lancé un projet pilote dans le contexte de l’hémato-oncologie afin de déterminer comment rembourser les tests NGS. Ces tests font désormais partie de la procédure standard et nous avons mis en place une convention NGS à part entière. Nous l’avons voulue flexible, afin que les nouvelles technologies puissent également être remboursées facilement. »
Des données relatives aux tests prédictifs individuels et aux tests NGS ont toujours été collectées via le registre PITTER. Quel est le rôle de ce registre ?
« En 2019, nous avons lié le remboursement des tests à l’enregistrement obligatoire des résultats des tests par les laboratoires dans le registre PITTER, accessible au public. Il ne s’agit pas d’informations génétiques spécifiques sur les patients, mais d’une interprétation générique. »
« Nous recueillons ainsi des données épidémiologiques sur les marqueurs pour l’ensemble de la Belgique ; une initiative assez unique en Occident. Il y a bien des données issues d’études cliniques, d’universités, etc., à petite échelle. Mais elles sont éparpillées et presque personne ne dispose de données nationales. Notre registre les recueille à présent en Belgique, et tout le monde en profitera. »
« Pratiquement aucun pays ne dispose de données épidémiologiques nationales sur les marqueurs. Avec le registre PITTER, nous les recueillons à présent en Belgique. »
« Nous obtenons des références nationales : les résultats des tests belges correspondent-ils à ce qui est publié au niveau international ? À plus petite échelle, nos laboratoires peuvent comparer leurs résultats à la référence belge : leurs résultats sont-ils conformes aux références nationales ou les laboratoires doivent-ils revoir leurs procédures de test en interne ? Les entreprises ont une meilleure idée du nombre de tests utilisés, du nombre de patients qui présentent telle ou telle mutation, etc. Elles peuvent donc réagir plus efficacement. »
« Et d’un point de vue politique, nous estimons mieux la valeur ajoutée réelle des traitements innovants. De quoi rectifier le tir si nécessaire. »
(lisez la suite sous la photo)

Sur quoi devrions-nous concentrer nos efforts en matière de companion diagnostics dans les mois et les années à venir ?
« Nous avons lancé le remboursement couplé des thérapies ciblées et des tests sur les marqueurs moléculaires en 2019, car c’est à ce niveau que les besoins étaient les plus criants. Depuis lors, nous continuons à chercher comment suivre l’évolution, par exemple en étendant les tests prédictifs à d’autres marqueurs : les marqueurs immunohistochimiques, mais aussi les marqueurs que nous détectons grâce à d’autres technologies médicales, comme l’imagerie médicale. »
« Toutes ces nouvelles technologies nécessitent que nous garantissions la qualité par divers moyens : normalisation, standardisation, directives, etc. Un patient pris en charge à Arlon doit avoir les mêmes options qu’un patient soigné à Ostende. »
« L’accent a été mis sur les marqueurs moléculaires durant cette législature. Entre-temps, nous cherchons comment suivre l'évolution, par exemple en étendant le remboursement lié à d'autres marqueurs. »
« Aujourd’hui, la Belgique s’en sort bien en termes d’accès aux traitements personnalisés, mais l’évaluation du projet NGS révèle aussi que tous les patients ne bénéficient pas des tests que la bonne pratique clinique voudrait qu’ils reçoivent. En d’autres termes, les soins personnalisés ne sont pas seulement une question de remboursement, il s’agit aussi de garantir l’encadrement et l’expertise nécessaires au sein des équipes soignantes. »
Comment faire en sorte que cet encadrement évolue aussi ?
« La clé du succès réside dans la concertation permanente avec l’ensemble des parties prenantes. La Commission de Médecine Personnalisée (ComPerMed) en est un excellent exemple. Il est primordial qu’un gouvernement sache ce qui se passe réellement sur le terrain. Et inversement, il est utile que le secteur ait une vision plus large des défis auxquels les décideurs politiques font face. La compréhension mutuelle est la base d’une véritable cocréation et de meilleures solutions. »